Revolutionizing Azelnidipine Production: A Mild, High-Yield Synthetic Route for Global Supply Chains
Revolutionizing Azelnidipine Production: A Mild, High-Yield Synthetic Route for Global Supply Chains
The pharmaceutical landscape for cardiovascular therapeutics is constantly evolving, driven by the need for more efficient and sustainable manufacturing processes for critical active pharmaceutical ingredients (APIs). A pivotal advancement in this domain is detailed in patent CN1752086A, which discloses a significantly improved method for preparing Azelnidipine, a potent third-generation dihydropyridine calcium channel blocker. Unlike traditional synthetic pathways that rely on harsh alkaline conditions and elevated temperatures, this novel approach utilizes a mild, one-pot condensation strategy involving 3-nitrobenzaldehyde, isopropyl acetoacetate, and a specialized guanidine derivative. By shifting the reaction environment from strongly basic to neutral or slightly acidic conditions, the process achieves exceptional control over impurity profiles, resulting in finished products with HPLC purity exceeding 99.5%. This technological breakthrough addresses long-standing challenges in yield optimization and operational safety, positioning it as a cornerstone for reliable azelnidipine intermediate supplier networks aiming to meet stringent global pharmacopeial standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
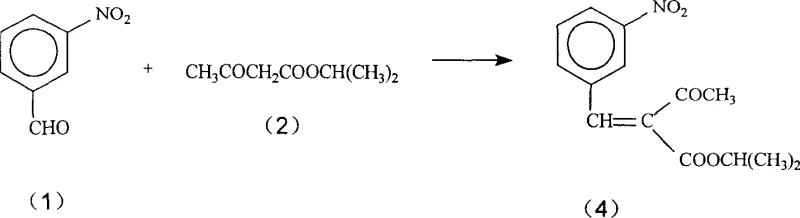
Historically, the industrial synthesis of Azelnidipine has been plagued by significant inefficiencies inherent to the legacy protocols established in early patents such as EP0266922. These conventional methods typically mandate the use of strong bases, including sodium methoxide, sodium hydroxide, or potassium hydroxide, coupled with high-temperature reflux in solvents like isopropanol. As illustrated in the reaction scheme below, this aggressive chemical environment often triggers undesirable side reactions, particularly affecting the sensitive guanidine functionality and the sterically hindered dihydropyridine ring system.
Practical implementation of these prior art methods frequently results in dismal actual yields, often hovering around 15%, which stands in stark contrast to the theoretical or literature-reported yields of over 85%. Furthermore, the crude product obtained from these harsh conditions is typically a brown viscous liquid with an HPLC purity of only approximately 95%, making subsequent purification to medicinal grade (>99.9%) extremely difficult and costly. The necessity for multiple recrystallization steps not only erodes overall mass balance but also introduces variability in particle size and polymorphic form, creating substantial bottlenecks for procurement managers seeking cost reduction in calcium channel blocker manufacturing.
The Novel Approach
In a decisive break from tradition, the methodology described in CN1752086A introduces a paradigm shift by conducting the Hantzsch-like condensation under mild, neutral, or slightly acidic conditions at temperatures ranging from 0°C to 60°C, with room temperature being optimal. This approach eliminates the need for corrosive strong bases, thereby preserving the integrity of the reactive amine and ester groups throughout the transformation. The process involves stirring the three key components—Compound (1), Compound (2), and Compound (3)—in a suitable solvent system, followed by a streamlined workup involving acidification, organic extraction, and sequential crystallization. This gentle reaction profile drastically reduces the formation of polymeric by-products and decomposition species, leading to isolated yields consistently above 70% and often reaching nearly 80% on a preparative scale. The simplicity of the operation, which avoids complex temperature cycling and hazardous reagent handling, translates directly into enhanced supply chain reliability and reduced operational expenditure for commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Mild Acid-Catalyzed Dihydropyridine Formation
To fully appreciate the technical superiority of this new route, one must analyze the mechanistic nuances that differentiate it from analogous syntheses like that of Nifedipine. In the synthesis of simpler dihydropyridines, such as Nifedipine, the condensation of 3-nitrobenzaldehyde with ethyl acetoacetate and beta-aminocrotonaldehyde proceeds relatively smoothly even under reflux in ethanol, as shown in the comparative reaction scheme below.
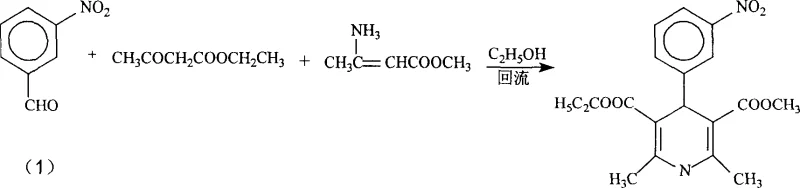
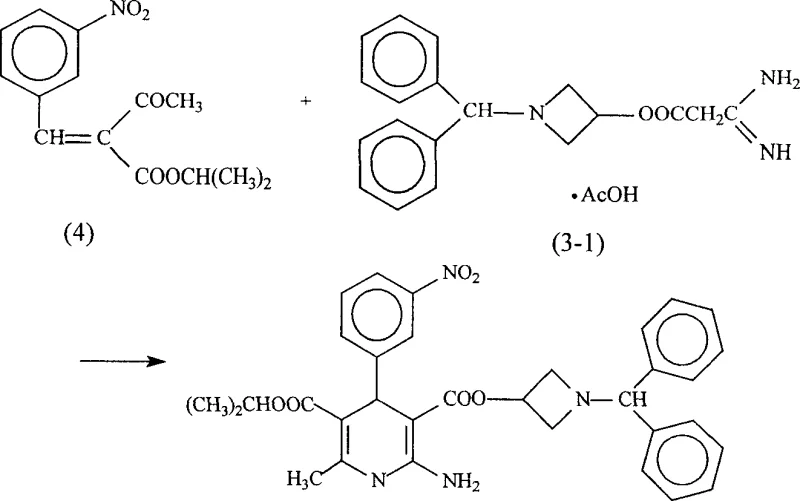
However, Azelnidipine presents a unique challenge due to the bulky 1-(diphenylmethyl)-3-azetidinyl group attached to the ester side chain and the presence of the guanidine moiety. The steric hindrance imposed by the diphenylmethyl group and the azetidine ring significantly slows down the nucleophilic attack required for ring closure. In conventional strong-base environments, the guanidine group is prone to hydrolysis or unwanted alkylation, leading to the observed low yields. The new method likely proceeds through a stabilized enamine intermediate, similar to Compound (4) shown in related literature, but avoids its isolation and the associated degradation risks.
By maintaining a slightly acidic or neutral pH, the reaction facilitates the reversible formation of the Schiff base between the aldehyde and the amine component without driving the equilibrium towards decomposition. The use of isopropyl acetoacetate (Compound 2), whose structure is depicted below, provides the necessary steric and electronic environment to favor the 1,4-dihydropyridine cyclization over competing Knoevenagel condensations. This precise control over the reaction trajectory ensures that the final product possesses the correct stereochemistry and substitution pattern required for biological activity, while minimizing the generation of regioisomers that are difficult to separate.
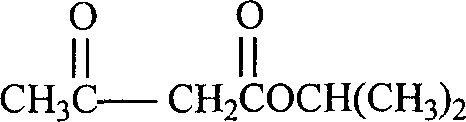
Furthermore, the workup procedure plays a critical mechanistic role in purity enhancement. The addition of dilute acid post-reaction serves to quench any residual basicity and protonate unreacted amines, rendering them water-soluble and easily removable during the aqueous wash. Subsequent extraction with ethyl acetate selectively pulls the neutral dihydropyridine product into the organic phase, leaving behind polar impurities. This logical sequence of pH-dependent extractions acts as a chemical filter, ensuring that the material entering the crystallization stage is already of high quality, thus reducing the burden on the final purification steps.
How to Synthesize Azelnidipine Efficiently
The practical execution of this improved synthesis is designed for seamless integration into existing multipurpose reactor setups commonly found in fine chemical facilities. The process begins with the suspension of the aldehyde, beta-keto ester, and guanidine salt in isopropanol, followed by agitation at ambient temperature for a period of 4 to 16 hours. Detailed standardized operating procedures regarding stoichiometry, mixing rates, and filtration parameters are essential for reproducibility. For a comprehensive step-by-step guide on implementing this technology in your facility, please refer to the technical protocol outlined below.
- Suspend 3-nitrobenzaldehyde, isopropyl acetoacetate, and the guanidine derivative salt in isopropanol and stir at room temperature (0-30°C) for 4-16 hours.
- Filter the reaction mixture, treat the filtrate with dilute acid, extract with ethyl acetate, and wash the organic layer with weak alkaline water followed by brine.
- Concentrate the organic phase, crystallize the residue from methanol to form the methanolate, and finally recrystallize from cyclohexane to obtain pure Azelnidipine.
Commercial Advantages for Procurement and Supply Chain Teams
For decision-makers overseeing the sourcing of cardiovascular intermediates, the adoption of this patented methodology offers profound strategic benefits that extend beyond mere chemical yield. The transition from a high-energy, hazardous process to a mild, ambient-temperature operation fundamentally alters the cost structure and risk profile of the supply chain. By eliminating the requirement for cryogenic cooling or high-temperature reflux, manufacturers can significantly reduce utility consumption and extend the lifespan of reactor vessels and gaskets that would otherwise suffer from corrosion by strong alkalis. This operational simplification directly contributes to substantial cost savings in API manufacturing, allowing for more competitive pricing models without compromising on quality margins.
- Cost Reduction in Manufacturing: The economic impact of this process is driven primarily by the elimination of expensive and hazardous reagents such as sodium methoxide and the reduction in energy intensity. Traditional methods require sustained heating and rigorous temperature control to manage exotherms, whereas this new route proceeds spontaneously at room temperature, drastically lowering electricity and steam costs. Additionally, the simplified workup reduces the volume of solvents and washing agents required, leading to lower waste disposal fees and a smaller environmental footprint. These cumulative efficiencies result in a leaner production cost base, enabling suppliers to offer high-purity azelnidipine at a more attractive price point.
- Enhanced Supply Chain Reliability: Supply continuity is often jeopardized by complex processes that are sensitive to minor deviations in raw material quality or operating conditions. The robustness of this mild synthesis makes it far more forgiving, reducing the incidence of batch failures and off-spec material that can disrupt downstream API production schedules. Since the reaction does not rely on strictly anhydrous conditions or specialized catalysts that may have long lead times, procurement teams can source raw materials from a broader range of qualified vendors. This flexibility mitigates the risk of supply shortages and ensures a steady flow of intermediates to meet the demands of global pharmaceutical markets.
- Scalability and Environmental Compliance: Scaling chemical processes from the laboratory to multi-ton production often reveals hidden challenges, particularly regarding heat transfer and mixing efficiency. The exothermic nature of strong-base catalyzed reactions poses significant safety risks at large scales, requiring expensive engineering controls. In contrast, the near-isothermal nature of this room-temperature reaction simplifies scale-up, allowing for larger batch sizes without proportional increases in safety infrastructure. Furthermore, the reduction in hazardous waste generation aligns with increasingly stringent environmental regulations, facilitating smoother regulatory approvals and enhancing the sustainability credentials of the manufacturing site.
Frequently Asked Questions (FAQ)
Understanding the technical specifics of this synthesis is crucial for stakeholders evaluating its potential for integration into their supply chains. The following questions address common inquiries regarding the reaction mechanism, purification strategies, and quality attributes of the final product. These answers are derived directly from the experimental data and technical disclosures within the patent documentation to provide accurate and actionable insights.
Q: Why does the new method achieve higher purity than conventional strong-base protocols?
A: Conventional methods utilize strong bases like sodium methoxide at high temperatures, which promote side reactions and degradation of the sensitive guanidine moiety. The new protocol operates under neutral or slightly acidic conditions at room temperature, significantly minimizing impurity formation and preserving the structural integrity of the dihydropyridine core.
Q: What are the critical solvent systems for optimizing yield in this synthesis?
A: While the reaction can proceed in various solvents, isopropanol is identified as the most suitable reaction medium due to its ability to dissolve reactants while facilitating the mild condensation. For extraction and purification, a combination of ethyl acetate and aqueous washes effectively removes unreacted starting materials, followed by methanol and cyclohexane for sequential crystallization to ensure >99% HPLC purity.
Q: How does this process impact commercial scalability for API manufacturers?
A: The elimination of high-temperature reflux and strong corrosive bases simplifies equipment requirements and reduces energy consumption. The robust nature of the room-temperature reaction allows for easier scale-up from kilogram to multi-ton production without the safety risks associated with exothermic strong-base additions, ensuring consistent supply continuity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Azelnidipine Supplier
As the global demand for effective antihypertensive therapies continues to rise, the need for a dependable source of high-quality Azelnidipine intermediates has never been more critical. NINGBO INNO PHARMCHEM stands at the forefront of this industry, leveraging advanced synthetic technologies like the one described in CN1752086A to deliver superior products. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistency and precision. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch meets or exceeds international pharmacopeial standards.
We invite you to collaborate with us to optimize your supply chain and reduce your overall manufacturing costs. Our technical experts are ready to provide a Customized Cost-Saving Analysis tailored to your specific production needs, demonstrating how our improved synthesis route can enhance your bottom line. Please contact our technical procurement team today to request specific COA data, route feasibility assessments, and sample quantities for your evaluation. Let us be your partner in delivering life-saving medications to patients worldwide through innovation and excellence.