Revolutionizing Azelnidipine Production: A One-Pot Strategy for High-Purity Calcium Channel Blockers
Revolutionizing Azelnidipine Production: A One-Pot Strategy for High-Purity Calcium Channel Blockers
The pharmaceutical industry continuously seeks robust synthetic routes that balance high purity with economic efficiency, particularly for complex calcium channel blockers like Azelnidipine. Patent CN100352818C introduces a transformative approach to synthesizing this critical antihypertensive agent, moving away from traditional multi-step protocols toward a streamlined one-pot condensation strategy. This innovation addresses long-standing challenges in yield consistency and impurity control, offering a compelling value proposition for reliable pharmaceutical intermediates supplier networks aiming to optimize their supply chains. By reacting 3-nitrobenzaldehyde, isopropyl acetoacetate, and the complex amine component simultaneously under mild conditions, the process eliminates the need for isolating unstable intermediates, thereby reducing operational complexity and enhancing overall process safety for commercial scale-up.
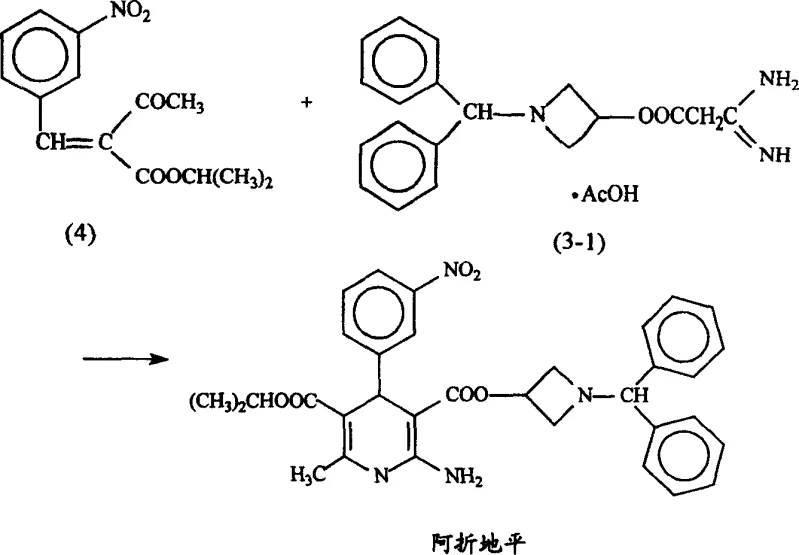
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Azelnidipine has relied on a sequential pathway where the dihydropyridine core is constructed first, followed by coupling with the bulky amine side chain under aggressive conditions. As illustrated in the conventional reaction scheme, this typically involves reacting pre-formed intermediate (4) with compound (3-1) in the presence of strong bases like sodium methoxide or potassium hydroxide at high reflux temperatures. This approach suffers from significant drawbacks, including the generation of numerous by-products due to the sensitivity of the guanidino group to strong alkalis. Practical implementation of these literature methods often results in dismal actual yields, reported as low as 15%, far below the theoretical expectations, alongside difficult purification processes that struggle to achieve the requisite 99.99% HPLC purity needed for regulatory approval. Furthermore, the high energy consumption associated with prolonged reflux and the handling of corrosive bases pose substantial safety and environmental liabilities for large-scale manufacturing facilities.
The Novel Approach
In stark contrast, the methodology disclosed in CN100352818C pioneers a direct three-component condensation that bypasses the isolation of the intermediate entirely. By combining compound (1), compound (2), and compound (3) in a single reactor, the process leverages a carefully balanced neutral or slightly acidic environment to drive the Hantzsch-like cyclization. This strategic shift not only simplifies the workflow by removing a distinct reaction vessel and filtration step but also fundamentally alters the reaction kinetics to favor the desired product over degradation pathways. The ability to conduct this transformation at temperatures ranging from 0°C to 60°C, preferably near room temperature, drastically reduces thermal stress on the molecule. Consequently, this novel route delivers a finished product with superior crystallinity and purity profiles, directly addressing the pain points of cost reduction in pharmaceutical intermediates manufacturing by minimizing waste and maximizing throughput without compromising quality standards.
Mechanistic Insights into One-Pot Hantzsch Condensation
The success of this one-pot synthesis hinges on the precise management of steric and electronic factors inherent to the reactants, particularly the bulky amine component shown in the structural diagram. Unlike simpler dihydropyridines such as nifedipine, Azelnidipine incorporates a complex 1-(diphenylmethyl)-3-azetidinyl group attached to a guanidine moiety, which introduces significant steric hindrance around the reaction center. In traditional basic conditions, this steric bulk combined with the nucleophilicity of the guanidine nitrogen can lead to polymerization or hydrolysis. However, the new protocol utilizes a micro-acidic or neutral medium, likely facilitating the formation of an enamine intermediate from the beta-keto ester and the aldehyde before the amine attacks, thus orchestrating the cyclization in a more controlled manner. This mechanistic nuance prevents the premature decomposition of the sensitive azetidine ring and ensures that the stereochemical integrity of the 1,4-dihydropyridine scaffold is maintained throughout the reaction trajectory.
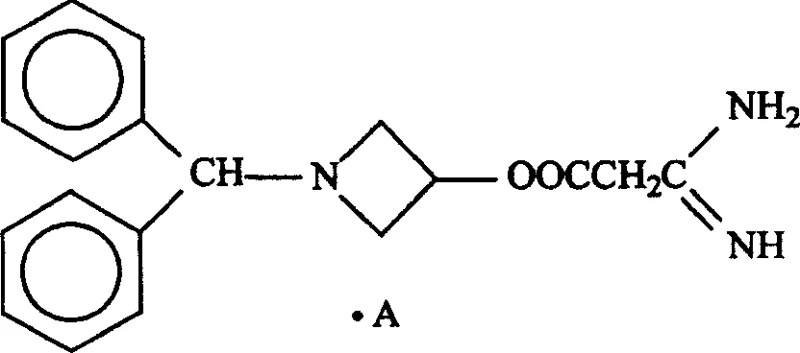
Furthermore, the impurity control mechanism is intrinsically linked to the workup procedure designed to quench unreacted starting materials without degrading the product. The process employs a specific sequence of acidification followed by organic extraction, which effectively partitions the basic amine impurities into the aqueous phase while retaining the neutral dihydropyridine product in the organic layer. This selective partitioning is critical because residual aldehyde or keto-ester can catalyze further unwanted reactions during concentration. By washing the organic layer with weak alkaline solutions like sodium bicarbonate after acid treatment, the process neutralizes any trapped acid catalysts that might promote oxidation of the dihydropyridine ring to the pyridine analog, a common degradation pathway. This meticulous attention to pH control during the isolation phase ensures that the final crude material enters the crystallization step with a purity profile that allows for easy upgrading to pharmaceutical grade through simple solvent exchange.
How to Synthesize Azelnidipine Efficiently
Implementing this optimized synthetic route requires strict adherence to the specified solvent systems and temperature controls to replicate the high yields reported in the patent examples. The procedure begins with the suspension of the three key precursors in isopropanol, where the reaction proceeds spontaneously over a period of 4 to 16 hours, eliminating the need for external heating sources in many embodiments. Following the reaction, the mixture is filtered to remove any insoluble particulates, and the filtrate is treated with dilute acetic acid to stabilize the system before extraction with ethyl acetate. The detailed standardized synthesis steps, including specific molar ratios, washing sequences, and crystallization parameters required for GMP compliance, are outlined in the guide below for technical teams evaluating process feasibility.
- Suspend compounds (1), (2), and (3) in isopropanol and stir at room temperature (0-30°C) for 4-16 hours to initiate the condensation reaction.
- Filter the reaction mixture, treat the filtrate with dilute acetic acid, and extract the product using ethyl acetate followed by washing with weak alkaline water and saturated brine.
- Concentrate the organic layer under reduced pressure below 60°C, crystallize the residue from methanol, and perform trans-crystallization using cyclohexane to obtain high-purity Azelnidipine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this one-pot methodology represents a significant opportunity to enhance margin structures and supply reliability. By consolidating what was previously a multi-stage operation into a single reactor train, manufacturers can drastically reduce the capital expenditure required for production equipment and the associated labor costs for monitoring multiple batch steps. The elimination of the intermediate isolation step not only saves time but also removes the yield losses typically incurred during filtration and drying of unstable intermediates, leading to a substantial increase in overall mass balance efficiency. Moreover, the use of common solvents like isopropanol and ethyl acetate, which are easily recoverable and recyclable, aligns with modern sustainability goals and reduces the total cost of ownership for solvent management systems in large-scale plants.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven primarily by the reduction in unit operations and the avoidance of expensive reagents. Traditional methods require stoichiometric amounts of strong bases and often necessitate cryogenic cooling or high-energy reflux, both of which inflate utility costs. In contrast, the new method operates near ambient temperature and utilizes catalytic or stoichiometric amounts of inexpensive acids for workup, significantly lowering the variable cost per kilogram. Additionally, the higher crude purity achieved directly from the reaction minimizes the need for extensive chromatographic purification or repeated recrystallizations, further driving down processing costs and increasing the effective capacity of existing manufacturing assets.
- Enhanced Supply Chain Reliability: Simplifying the synthetic route inherently reduces the number of potential failure points in the supply chain, ensuring more consistent delivery schedules for downstream API manufacturers. The robustness of the reaction against minor fluctuations in temperature and mixing rates means that batch-to-batch variability is minimized, a critical factor for maintaining qualified supplier status with major pharmaceutical clients. Furthermore, the raw materials required—3-nitrobenzaldehyde and isopropyl acetoacetate—are commodity chemicals with stable global availability, mitigating the risk of supply disruptions that often plague specialized intermediates. This stability allows for better inventory planning and reduces the need for excessive safety stock holdings.
- Scalability and Environmental Compliance: From an environmental and scaling perspective, the absence of harsh alkaline waste streams simplifies effluent treatment and reduces the burden on wastewater processing facilities. The process generates less hazardous waste, making it easier to comply with increasingly stringent environmental regulations in key manufacturing regions. Scaling this reaction from pilot to commercial tonnage is straightforward because the exotherm is manageable and the reaction does not rely on difficult-to-control high-temperature reflux conditions. This ease of scale-up ensures that commercial scale-up of complex pharmaceutical intermediates can be achieved rapidly to meet market demand without requiring extensive re-engineering of the process parameters.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method, providing clarity for R&D and procurement stakeholders evaluating this technology. These insights are derived directly from the experimental data and comparative analysis presented in the patent documentation, ensuring accuracy and relevance for decision-making processes.
Q: How does the new one-pot method improve yield compared to conventional methods?
A: The conventional method involves a two-step process with harsh alkaline conditions and high-temperature reflux, often resulting in actual yields as low as 15% due to side reactions. The new one-pot method operates under neutral or slightly acidic conditions at lower temperatures (0-60°C), significantly reducing decomposition and achieving yields exceeding 70% with HPLC purity above 99%.
Q: What are the critical solvent systems used in this optimized synthesis?
A: The primary reaction solvent is isopropanol, which effectively suspends the reactants. For extraction and purification, the process utilizes ethyl acetate, chloroform, or dichloromethane. The final purification involves a methanol crystallization step followed by trans-crystallization in cyclohexane to remove solvent residues and ensure pharmaceutical-grade quality.
Q: Why is avoiding strong base catalysts significant for Azelnidipine stability?
A: Azelnidipine contains sensitive functional groups, including a guanidino moiety and an azetidine ring, which are prone to degradation under strong alkaline conditions (e.g., sodium methoxide). By shifting to a neutral or micro-acidic environment, the new method preserves the structural integrity of these groups, minimizing impurity formation and simplifying downstream purification.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Azelnidipine Supplier
At NINGBO INNO PHARMCHEM, we understand that the transition to a more efficient synthetic route requires a partner with deep technical expertise and proven manufacturing capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this one-pot process are fully realized in a GMP-compliant environment. We maintain stringent purity specifications and operate rigorous QC labs equipped to verify the low impurity profiles and specific crystalline forms required for global regulatory filings, giving our partners confidence in the quality and consistency of every batch supplied.
We invite you to engage with our technical procurement team to discuss how this optimized Azelnidipine synthesis can be integrated into your supply chain to drive value. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the potential economic impact specific to your volume requirements. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project timelines, ensuring a seamless path from development to commercial success.