Revolutionizing Tigecycline Production: A Direct 6-Step Synthesis from Demethyl Aureomycin
Introduction to Novel Tigecycline Synthesis Technology
The pharmaceutical industry continuously seeks more efficient pathways for producing complex antibiotics, and Patent CN109824539B presents a significant breakthrough in the manufacturing of Tigecycline, a potent glycylcycline antibiotic. This intellectual property discloses a novel method for synthesizing Tigecycline directly from Demethyl Aureomycin (DMC), bypassing the traditional and cumbersome intermediate synthesis of Minocycline. By streamlining the synthetic route into six concise steps—including nitration, catalytic reduction, acylation, and methylation—this technology addresses critical bottlenecks in yield, purity, and operational safety. For R&D directors and procurement strategists, this represents a pivotal shift towards more sustainable and cost-effective API production, leveraging the abundant availability of fermentation-derived DMC to create high-value therapeutic agents with reduced environmental impact.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of Tigecycline has relied heavily on Minocycline as a starting material, a pathway fraught with inefficiencies and safety hazards. As illustrated in prior art such as patent CN200710171556.7, traditional routes often necessitate up to eight reaction steps, involving complex protection and deprotection strategies that introduce significant yield losses at every stage. These conventional methods frequently employ toxic reagents, such as hydrogen chloride gas for deprotection, which imposes severe requirements on equipment corrosion resistance and operator safety protocols. Furthermore, the reliance on solvents like dimethylformamide (DMF) and diethyl ether creates substantial downstream processing challenges; DMF is difficult to recover due to its high boiling point, while diethyl ether poses significant explosion risks and storage difficulties, rendering these processes less ideal for modern, large-scale green chemistry standards.
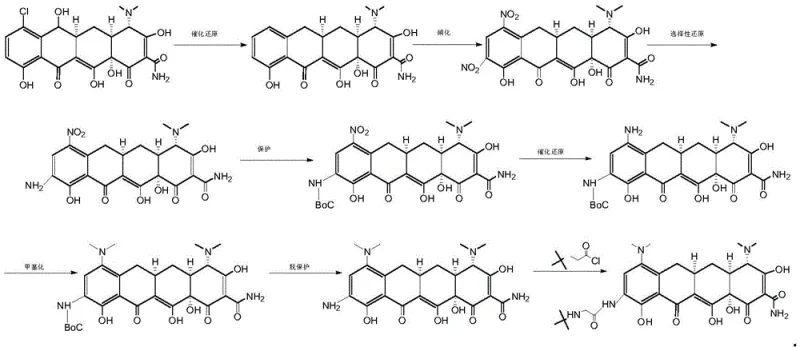
The Novel Approach
In stark contrast, the methodology outlined in CN109824539B offers a streamlined, six-step trajectory that directly transforms Demethyl Aureomycin into Tigecycline, effectively cutting out the intermediate Minocycline synthesis entirely. This innovative route eliminates the need for protective group chemistry, thereby removing the associated deprotection steps and the hazardous reagents they require. The process utilizes a robust sequence of nitration and catalytic hydrogenation reactions that are easier to control and monitor, significantly reducing the formation of impurities that typically plague selective reduction steps in older methodologies. By adopting safer solvent systems like ethyl acetate and n-butanol for crystallization, this approach not only enhances the purity profile of the final API but also simplifies solvent recovery, making it a superior choice for cost reduction in pharmaceutical manufacturing.
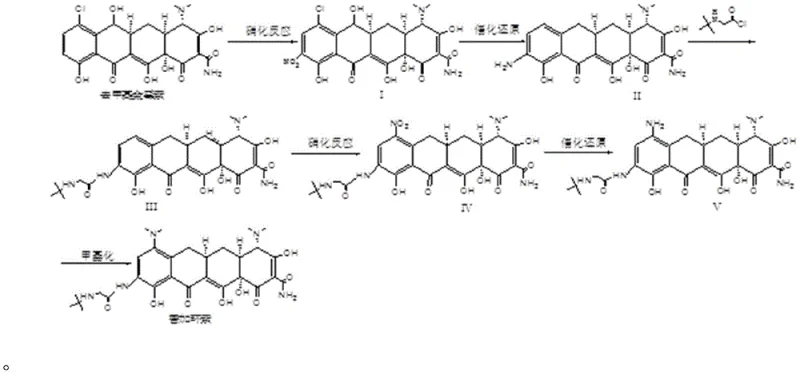
Mechanistic Insights into Catalytic Hydrogenation and Nitration
The core of this synthetic innovation lies in the precise control of electrophilic aromatic substitution and heterogeneous catalysis. The initial nitration of Demethyl Aureomycin is conducted under strictly controlled low-temperature conditions (3-6°C) using potassium nitrate in concentrated sulfuric acid, ensuring regioselective introduction of the nitro group at the 9-position of the tetracycline ring system without degrading the sensitive molecular scaffold. Subsequent catalytic reduction steps utilize palladium on carbon (Pd/C) under hydrogen pressure exceeding 4.0 MPa, a condition optimized to reduce the nitro groups to amines efficiently while preserving the stereochemical integrity of the multiple chiral centers inherent to the tetracycline core. This mechanistic precision is crucial for maintaining the biological activity of the final antibiotic, as any epimerization at the C4 or C4a positions could render the product inactive or toxic.
Furthermore, the final methylation step employs a reductive amination strategy, where the intermediate amine reacts with formaldehyde or paraformaldehyde in the presence of a hydrogenation catalyst. This one-pot transformation installs the critical dimethylamino group required for Tigecycline's enhanced ribosomal binding affinity. The mechanism avoids the use of harsh alkylating agents that could lead to over-alkylation or side reactions on other nucleophilic sites within the molecule. By fine-tuning the pH during the workup and crystallization phases, the process ensures that the final product precipitates with high chemical purity, effectively minimizing the burden on downstream purification units and ensuring a consistent impurity profile that meets stringent regulatory specifications for antibiotic APIs.
How to Synthesize Tigecycline Efficiently
The synthesis of Tigecycline via this novel route involves a carefully orchestrated sequence of reactions starting from the fermentation product Demethyl Aureomycin. The process begins with a low-temperature nitration to functionalize the aromatic ring, followed by a high-pressure catalytic reduction to generate the key amino intermediate. This intermediate is then coupled with a glycyl moiety using tert-butylamine acetyl chloride hydrochloride, followed by a second cycle of nitration and reduction to finalize the side chain architecture. The detailed standardized synthesis steps, including specific molar ratios, temperature gradients, and pressure settings for each reactor stage, are provided in the technical guide below to ensure reproducibility and safety during scale-up operations.
- Perform nitration on Demethyl Aureomycin using potassium nitrate and sulfuric acid at low temperature to obtain Product I.
- Execute catalytic reduction of Product I using Pd/C and hydrogen pressure to generate the amino intermediate Product II.
- React Product II with tert-butylamine acetyl chloride hydrochloride, followed by a second nitration and reduction cycle to install the glycyl side chain.
- Conclude with a methylation reaction using formaldehyde and hydrogenation to yield the final Tigecycline crude product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthesis route translates into tangible strategic advantages regarding cost stability and supply continuity. By reducing the total number of synthetic steps from eight to six, the process inherently minimizes the cumulative yield loss typically associated with multi-step organic synthesis, leading to a higher overall output of finished API per unit of starting material. This step economy directly correlates to a significant reduction in raw material consumption and waste generation, driving down the variable costs of production without compromising on the quality or potency of the final pharmaceutical product.
- Cost Reduction in Manufacturing: The elimination of protection and deprotection steps removes the need for expensive reagents like Boc-anhydrides and toxic gases like hydrogen chloride, which require specialized scrubbing and containment infrastructure. Additionally, replacing difficult-to-recycle solvents like DMF with more volatile and recoverable alternatives like ethyl acetate reduces utility costs associated with distillation and waste treatment. These cumulative efficiencies result in substantial cost savings in API manufacturing, allowing for more competitive pricing in the global generic antibiotic market.
- Enhanced Supply Chain Reliability: Demethyl Aureomycin is a direct fermentation product that is widely available and stable, unlike Minocycline which requires further chemical processing before it can be used as a starting material. Relying on a more upstream, abundant raw material mitigates the risk of supply bottlenecks that can occur when depending on semi-synthetic intermediates. This robustness ensures a more reliable API intermediate supplier capability, safeguarding production schedules against fluctuations in the availability of complex precursors.
- Scalability and Environmental Compliance: The process is designed with industrial scalability in mind, utilizing heterogeneous catalysts that can be easily filtered and recycled, and avoiding cryogenic conditions that are energy-intensive to maintain on a metric-ton scale. The absence of highly toxic reagents and the use of greener solvent systems align with increasingly strict environmental regulations, reducing the regulatory burden and potential liability associated with hazardous waste disposal, thus facilitating smoother commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Tigecycline synthesis technology. These answers are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on how this method outperforms legacy processes in terms of safety, yield, and operational simplicity for industrial partners.
Q: What are the primary advantages of using Demethyl Aureomycin over Minocycline for Tigecycline synthesis?
A: Using Demethyl Aureomycin directly eliminates the need for the complex dechlorination and dehydroxylation steps required to first synthesize Minocycline. This shortens the overall route from 8 steps to 6 steps, significantly reducing material loss and processing time while avoiding the use of toxic reagents associated with the intermediate conversion.
Q: How does this novel method address safety concerns found in prior art patents?
A: Unlike previous methods (e.g., CN200710171556.7) that utilize toxic hydrogen chloride gas for deprotection and hazardous solvents like diethyl ether, this novel process operates without protection/deprotection cycles. It utilizes safer crystallization solvents like ethyl acetate and n-butanol, drastically improving operational safety and environmental compliance.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the process is specifically designed for scalability. It avoids difficult-to-recover high-boiling solvents like DMF and eliminates the need for cryogenic conditions beyond standard ice-salt baths. The use of heterogeneous catalysts like Pd/C allows for easy filtration and recycling, making it highly viable for commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tigecycline Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this streamlined synthesis route for the global antibiotic market. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patent are fully realized in a GMP-compliant manufacturing environment. Our rigorous QC labs and stringent purity specifications guarantee that every batch of Tigecycline produced meets the highest international pharmacopoeia standards, providing our partners with a secure and high-quality supply of this critical medicine.
We invite forward-thinking pharmaceutical companies to collaborate with us to leverage this advanced technology for their supply chains. By contacting our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to obtain specific COA data and route feasibility assessments, ensuring that your transition to this more efficient manufacturing process is seamless, compliant, and commercially advantageous.