Advanced Synthesis of Diarylethene-Based LSD1/HDAC Dual-Target Inhibitors for Oncology
Advanced Synthesis of Diarylethene-Based LSD1/HDAC Dual-Target Inhibitors for Oncology
The landscape of epigenetic therapy is rapidly evolving, with dual-target inhibitors emerging as a superior strategy to overcome resistance mechanisms often seen with single-agent therapies. Patent CN111592487B discloses a novel class of diarylethene-based compounds containing a hydroxamic acid group, specifically designed to simultaneously inhibit Lysine-Specific Demethylase 1 (LSD1) and Histone Deacetylases (HDACs). This technological breakthrough represents a significant leap forward in the development of next-generation anticancer agents, particularly for difficult-to-treat malignancies such as colon and gastric cancers. The structural versatility of these molecules, defined by the general formula I-5, allows for extensive SAR exploration to optimize potency and pharmacokinetic profiles.
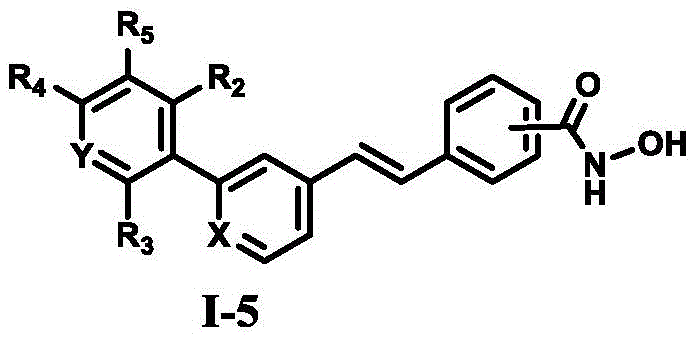
For pharmaceutical companies seeking a reliable pharmaceutical intermediates supplier, understanding the robustness of the underlying synthetic chemistry is paramount. The disclosed compounds exhibit nanomolar to micromolar inhibitory activity against both LSD1 and HDAC1/6, with several derivatives demonstrating superior in vitro antitumor activity compared to the marketed drug SAHA. This document provides a deep technical analysis of the synthesis, mechanism, and commercial viability of these high-value epigenetic modulators, highlighting opportunities for cost reduction in anticancer drug manufacturing through efficient process design.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to developing epigenetic modulators often rely on linear synthesis routes that suffer from poor atom economy and low overall yields, particularly when introducing complex pharmacophores like hydroxamic acids late in the sequence. Conventional HDAC inhibitors often lack selectivity, leading to dose-limiting toxicities, while standalone LSD1 inhibitors may fail to induce sufficient gene re-expression to trigger apoptosis in solid tumors. Furthermore, many existing synthetic pathways utilize expensive transition metal catalysts that are difficult to remove to ppm levels required for GMP production, or they require harsh reaction conditions that limit the scope of compatible functional groups. These inefficiencies create bottlenecks in the commercial scale-up of complex oncology intermediates, driving up costs and extending lead times for clinical trial material.
The Novel Approach
The methodology described in CN111592487B employs a convergent synthetic strategy that effectively decouples the construction of the diarylethene backbone from the installation of the zinc-binding hydroxamic acid warhead. By utilizing a Horner-Wadsworth-Emmons (HWE) olefination followed by a Suzuki-Miyaura cross-coupling, the process ensures high stereocontrol (E-isomer) and excellent functional group tolerance. This modular approach allows for the rapid generation of diverse analogues by simply swapping commercially available boronic acids or aldehydes. The final conversion to the hydroxamic acid is performed under mild conditions, preserving the integrity of the sensitive alkene linkage. This strategic design not only improves overall yield but also simplifies purification, directly addressing the need for high-purity epigenetic modulators in early-stage drug discovery.
Mechanistic Insights into Dual-Target Epigenetic Modulation
The biological efficacy of these compounds stems from their ability to engage two distinct epigenetic targets within the same molecular framework. The hydroxamic acid moiety acts as a potent zinc-binding group (ZBG), chelating the zinc ion located at the bottom of the HDAC enzyme active site tunnel, thereby blocking deacetylation and leading to hyperacetylation of histones. Simultaneously, the diarylethene scaffold interacts with the FAD-binding pocket of LSD1, preventing the oxidative demethylation of histone H3K4. This dual inhibition disrupts the co-repressor complexes (such as NuRD and CoREST) where LSD1 and HDACs physically interact, creating a synergistic effect that reactivates silenced tumor suppressor genes more effectively than either mechanism alone.
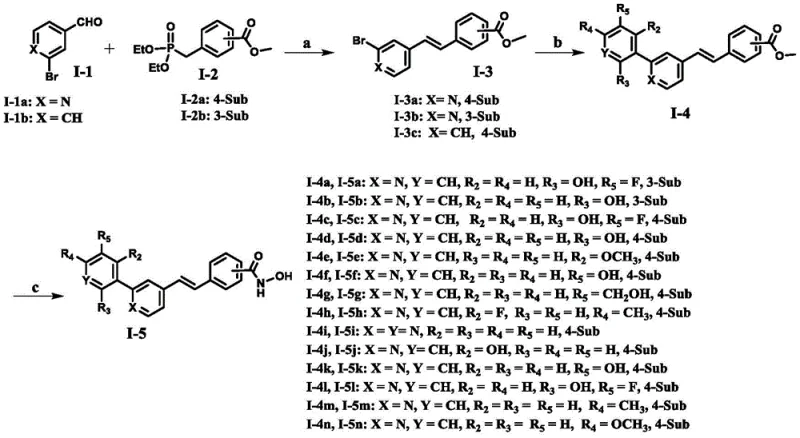
From a process chemistry perspective, the synthetic route minimizes impurity formation through careful control of reaction parameters. The HWE reaction utilizes strong bases like potassium tert-butoxide in DMF at room temperature, which favors the thermodynamic E-alkene product, reducing the burden of isomeric separation. The subsequent Suzuki coupling employs standard palladium catalysts like Pd(PPh3)4 with inorganic bases in a biphasic toluene/water system, a robust method known for scalability. The final aminolysis step uses freshly prepared hydroxylamine potassium salt, ensuring complete conversion of the ester without hydrolyzing the amide bond. This mechanistic understanding is critical for reducing lead time for high-purity pharmaceutical intermediates by predicting and mitigating potential process deviations during technology transfer.
How to Synthesize Diarylethene LSD1/HDAC Inhibitors Efficiently
The synthesis of these dual-target inhibitors is achieved through a streamlined three-step sequence that balances efficiency with flexibility. The process begins with the condensation of a substituted benzaldehyde and a phosphonate ester to form the stilbene core, followed by palladium-catalyzed cross-coupling to introduce the diverse aryl or heteroaryl substituents. The final step involves the conversion of the methyl ester to the bioactive hydroxamic acid. This route has been validated across multiple examples (I-5a to I-5n), demonstrating consistent yields and purity profiles suitable for biological evaluation. The detailed standardized synthesis steps are outlined in the guide below.
- Perform Horner-Wadsworth-Emmons reaction between substituted benzaldehyde and phosphonate ester using strong base in DMF.
- Execute Suzuki-Miyaura coupling between the bromo-alkene intermediate and substituted boronic acid using Palladium catalyst.
- Convert the methyl ester to hydroxamic acid using hydroxylamine hydrochloride and potassium hydroxide in methanol.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic platform offers tangible logistical and economic benefits derived from its chemical design. The reliance on commodity starting materials such as substituted benzaldehydes and boronic acids ensures a stable supply chain with multiple global sources, mitigating the risk of single-supplier dependency. The reaction conditions are generally mild, avoiding the need for specialized high-pressure equipment or cryogenic temperatures, which simplifies facility requirements and reduces energy consumption. Furthermore, the use of standard purification techniques like silica gel chromatography and recrystallization facilitates easy technology transfer to contract manufacturing organizations (CMOs).
- Cost Reduction in Manufacturing: The convergent nature of the synthesis significantly lowers the cost of goods sold (COGS) by maximizing the value of each synthetic step. By delaying the introduction of the more expensive hydroxamic acid functionality until the final step, the process avoids wasting valuable reagents on intermediates that might fail quality control earlier in the sequence. Additionally, the elimination of exotic reagents in favor of common bases and catalysts reduces raw material expenses. The high stereoselectivity of the HWE reaction minimizes the loss of material due to isomeric impurities, further enhancing overall process efficiency and yield.
- Enhanced Supply Chain Reliability: The modular design of the molecule allows for rapid adaptation to raw material shortages. If a specific boronic acid becomes unavailable, the synthetic route can accommodate alternative coupling partners with minimal re-optimization. The robustness of the Suzuki coupling reaction, which tolerates a wide range of functional groups and water content, ensures consistent batch-to-batch reproducibility. This reliability is crucial for maintaining continuous supply for clinical trials and prevents costly delays associated with process failures or impurity spikes.
- Scalability and Environmental Compliance: The synthetic pathway is inherently scalable, having been demonstrated on multi-gram scales in the patent examples with consistent results. The solvents used, such as DMF, toluene, and dichloromethane, are standard industrial solvents with well-established recovery and recycling protocols, aiding in environmental compliance and waste reduction. The absence of highly toxic reagents or unstable intermediates simplifies safety management and regulatory filing, accelerating the path from laboratory synthesis to commercial production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these LSD1/HDAC dual-target inhibitors. These answers are derived directly from the experimental data and specifications provided in the patent documentation, ensuring accuracy for R&D and procurement decision-making. Understanding these details is essential for evaluating the feasibility of integrating these intermediates into your drug discovery pipeline.
Q: What is the primary advantage of this dual-target inhibitor structure?
A: The diarylethene scaffold combined with a hydroxamic acid group allows for simultaneous inhibition of LSD1 and HDAC enzymes, offering synergistic antitumor effects superior to single-target agents like SAHA.
Q: Are the starting materials for this synthesis commercially available?
A: Yes, the key building blocks such as substituted benzaldehydes, diethyl benzylphosphonates, and various boronic acids are widely available commodity chemicals, ensuring supply chain stability.
Q: How does the selectivity of these compounds compare to MAO enzymes?
A: Experimental data indicates high selectivity for LSD1 over homologous MAO-A and MAO-B enzymes, with selectivity indices greater than 36-fold, reducing potential off-target side effects.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Diarylethene Inhibitor Supplier
NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis for complex epigenetic modulators, leveraging deep expertise in palladium-catalyzed cross-couplings and hydroxamate chemistry. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from milligram-scale screening to kilogram-scale clinical supply. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications required for oncology drug candidates, including precise control over residual metals and isomeric ratios.
We invite you to collaborate with our technical team to explore how this dual-target technology can accelerate your oncology portfolio. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our optimized processes can deliver high-quality intermediates with superior lead times and cost efficiency.