Advanced Four-Step Synthesis of Fluconazole: Enhancing Purity and Commercial Scalability
The pharmaceutical industry continuously seeks robust synthetic pathways that balance high purity with economic viability, particularly for broad-spectrum antifungal agents like Fluconazole. Patent CN101891693B introduces a transformative four-step synthesis method that fundamentally alters the production landscape for this critical Active Pharmaceutical Ingredient (API). Unlike traditional routes that rely on costly epoxidation reagents, this novel approach leverages a ketal protection strategy starting from 1,3-dibromoacetone. The process is characterized by its operational simplicity, mild reaction conditions, and the strategic elimination of high-priced raw materials, positioning it as a superior choice for cost reduction in pharmaceutical intermediates manufacturing. By integrating ionic liquid catalysis in the substitution step, the method achieves enhanced selectivity and yield, addressing long-standing challenges in impurity control.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of Fluconazole has been dominated by routes involving the reaction of m-difluorobenzene with chloroacetyl chloride, followed by epoxidation using Trimethylsulfoxonium Iodide or Trimethylsulfonium Iodide. While established, these conventional methods suffer from significant economic and technical drawbacks. The reliance on sulfonium salts introduces a major cost burden due to the high price of these reagents, which directly inflates the production cost of the finished drug. Furthermore, the epoxidation step often exhibits suboptimal yields and generates complex by-product profiles that complicate downstream purification. Alternative methods attempting direct Grignard reactions on 1,3-dichloroacetone have also struggled with poor selectivity and low overall yields, rendering them less attractive for large-scale commercial adoption where consistency and margin are paramount.
The Novel Approach
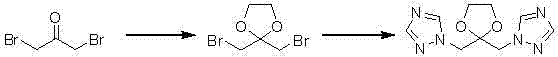
The methodology disclosed in CN101891693B circumvents these bottlenecks by initiating the synthesis with 1,3-dibromoacetone, a more economically accessible starting material. The core innovation lies in the initial ketalization to form 2,2-bis(bromomethyl)-1,3-dioxolane, which serves as a stable scaffold for subsequent functionalization. This route completely bypasses the need for expensive sulfonium ylides, replacing them with a efficient nucleophilic substitution facilitated by ionic liquids. The process flow is linear and logical, moving from protection to substitution, then deprotection, and finally carbon-carbon bond formation via Grignard chemistry. This structural redesign not only lowers the raw material cost baseline but also simplifies the workflow, making it an ideal candidate for reliable fluconazole intermediate supplier networks seeking to optimize their portfolios.
Mechanistic Insights into Ionic Liquid-Catalyzed Substitution and Grignard Addition
The heart of this synthetic advancement is the second step, where the dibromo-ketal intermediate undergoes double nucleophilic substitution with 1H-1,2,4-triazole. In this critical transformation, the patent specifies the use of ionic liquids, such as 1-ethyl-3-methylimidazolium hydrogen sulfate, acting as both solvent and catalyst. This dual functionality enhances the solubility of the triazole and stabilizes the transition state, leading to superior reaction kinetics compared to traditional phase-transfer catalysts. The reaction proceeds smoothly at moderate temperatures around 35°C in dichloromethane, minimizing thermal degradation risks. Following this, the dioxolane protecting group is removed via acid hydrolysis using hydrochloric acid at 100°C, regenerating the ketone functionality essential for the final coupling. The sequence concludes with the addition of a 3,5-difluorophenyl Grignard reagent to the ketone, forming the tertiary alcohol center of Fluconazole with high stereochemical integrity.
Impurity control is rigorously managed through the crystallization steps embedded within the process. For instance, the intermediate 1,3-bis(1H-1,2,4-triazole-1-yl)acetone is purified via recrystallization from ethanol, ensuring that only the highest quality material enters the final Grignard step. This is crucial because impurities at the ketone stage can lead to difficult-to-remove side products in the final API. The final Fluconazole product is isolated as a white crystalline powder after recrystallization from an isopropanol-water system, achieving a melting point range of 138-141°C consistent with high-purity standards. The detailed mechanistic understanding allows for precise tuning of reaction parameters, such as the drop rate of the Grignard reagent, to maintain temperatures below 50°C and prevent exothermic runaways.
How to Synthesize Fluconazole Efficiently
Implementing this synthesis requires careful attention to the sequential addition of reagents and temperature control across the four distinct stages. The process begins with the azeotropic removal of water during ketalization to drive the equilibrium forward, followed by a base-mediated substitution where the choice of ionic liquid is critical for maximizing yield. The hydrolysis step demands robust acid handling capabilities, while the final Grignard reaction requires strictly anhydrous conditions to ensure reagent efficacy. Detailed standardized operating procedures for each unit operation, including specific work-up protocols like pH adjustment and solvent swapping, are essential for reproducibility. The following guide outlines the critical operational parameters derived from the patent examples to assist technical teams in process validation.
- Perform ketalization of 1,3-dibromoacetone with ethylene glycol using p-toluenesulfonic acid catalyst in toluene at 100°C.
- Execute nucleophilic substitution with 1H-1,2,4-triazole using ionic liquid catalyst and base in dichloromethane.
- Conduct acid hydrolysis of the dioxolane intermediate followed by Grignard reaction with 3,5-difluorobromobenzene.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the shift to this synthetic route offers tangible strategic benefits beyond mere technical feasibility. The primary advantage is the drastic simplification of the raw material basket; by eliminating the dependency on specialized and expensive sulfonium salts, the supply chain becomes more resilient to market fluctuations. The use of commodity chemicals like 1,3-dibromoacetone, ethylene glycol, and common solvents ensures that sourcing is straightforward and competitive. This stability translates directly into cost reduction in pharmaceutical intermediates manufacturing, allowing for better margin management in the final drug product. Furthermore, the mild reaction conditions reduce energy consumption and equipment wear, contributing to a lower total cost of ownership for the production facility.
- Cost Reduction in Manufacturing: The elimination of high-cost reagents like Trimethylsulfoxonium Iodide removes a significant line item from the bill of materials. Additionally, the high yields reported in the patent examples, such as the 91.9% yield in the initial ketalization step and 88.0% in the triazole substitution, minimize waste and maximize throughput. The ability to use crude intermediates directly in subsequent steps without extensive purification further reduces processing time and solvent usage, driving down operational expenditures significantly.
- Enhanced Supply Chain Reliability: Sourcing 1,3-dibromoacetone and 3,5-difluorobromobenzene is far more reliable than securing specialized epoxidation agents, which may have limited suppliers. This diversification of the supply base mitigates the risk of production stoppages due to raw material shortages. The robustness of the chemistry also means that the process is less sensitive to minor variations in feedstock quality, ensuring consistent output even when supply chains are under stress, thereby reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The process operates at atmospheric pressure and moderate temperatures, utilizing standard glass-lined or stainless steel reactors found in most multipurpose plants. This ease of scale-up facilitates the commercial scale-up of complex pharmaceutical intermediates without requiring capital-intensive new infrastructure. Moreover, the reduced use of hazardous reagents and the potential for solvent recovery align with modern environmental, health, and safety (EHS) standards, simplifying regulatory compliance and waste disposal logistics.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Fluconazole synthesis route. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation. Understanding these nuances is vital for R&D teams evaluating technology transfer and for procurement officers assessing supplier capabilities. The answers reflect the practical realities of scaling this chemistry from the laboratory to multi-ton production.
Q: What are the key advantages of this Fluconazole synthesis route over traditional methods?
A: This route avoids the use of expensive Trimethylsulfoxonium Iodide required in epoxide-based methods. It utilizes readily available 1,3-dibromoacetone and employs ionic liquids to enhance selectivity and yield in the substitution step, significantly reducing raw material costs.
Q: How does the use of ionic liquids impact the reaction efficiency?
A: The patent specifies using ionic liquids such as 1-ethyl-3-methylimidazolium hydrogen sulfate as phase-transfer catalysts. This substitution simplifies the operating process, shortens reaction times, and improves the selectivity of the triazole substitution step compared to traditional phase-transfer catalysts.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the reaction conditions are mild (temperatures ranging from 35°C to 100°C) and utilize conventional unit operations like reflux, distillation, and crystallization. The avoidance of specialized high-pressure equipment and the use of common solvents like toluene and THF make it highly scalable for industrial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fluconazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic routes like the one described in CN101891693B to maintain competitiveness in the global API market. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this ionic liquid-catalyzed process are fully realized in practice. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Fluconazole intermediate meets the highest international pharmacopoeia standards. Our commitment to quality ensures that your downstream formulation processes remain uninterrupted and compliant.
We invite you to collaborate with us to leverage this cost-effective technology for your supply chain. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our optimized manufacturing capabilities can enhance your product margins and supply security.