Advanced Salt-Formation Strategy for High-Purity Clevidipine Butyrate Manufacturing
The pharmaceutical industry continuously seeks robust synthetic routes for potent cardiovascular agents, and the ultra-short-acting calcium channel blocker clevidipine butyrate represents a critical asset for perioperative blood pressure management. Patent CN102001992A introduces a transformative methodology for preparing this active pharmaceutical ingredient (API) intermediate, specifically addressing the longstanding purification challenges associated with dihydropyridine derivatives. The core innovation lies in a strategic salt-formation and recrystallization protocol that bypasses the traditionally cumbersome column chromatography steps. By converting the key mono-acid intermediate into a stable metal salt, manufacturers can achieve exceptional purity levels exceeding 99.5% through simple crystallization techniques. This technical breakthrough not only enhances the chemical profile of the final product but also aligns perfectly with the rigorous demands of modern Good Manufacturing Practice (GMP) environments, offering a reliable pathway for producing high-purity clevidipine butyrate suitable for injectable formulations.
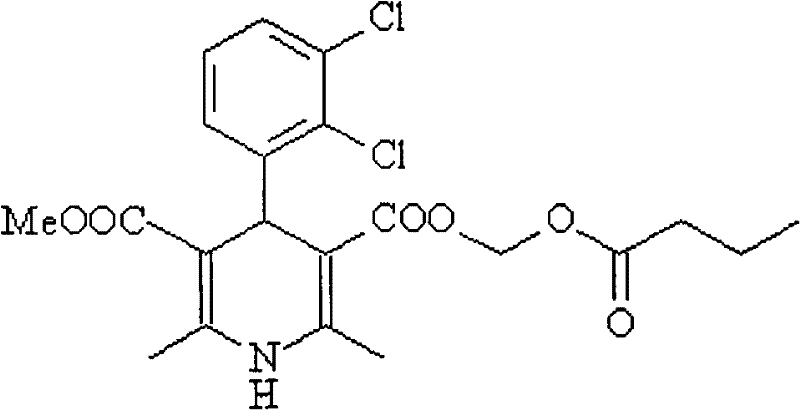
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of clevidipine analogues has relied heavily on the Hantzsch condensation reaction, a classic method for constructing the 1,4-dihydropyridine ring system. However, this approach is plagued by significant selectivity issues, often yielding a complex mixture of products including the desired mono-ester/mono-acid alongside symmetrical di-ester and di-acid impurities. In many documented procedures, these symmetrical byproducts can constitute anywhere from 2% to 25% of the crude reaction mass, creating a severe purification burden. The structural similarity between the target mono-acid intermediate and these symmetric impurities makes separation via standard extraction or simple washing nearly impossible. Consequently, the industry standard has long depended on flash column chromatography to isolate the pure intermediate. For large-scale manufacturing, column chromatography is economically and operationally prohibitive due to the massive volumes of solvents required, the high cost of stationary phases like silica gel, and the low throughput inherent in batch chromatographic processes. Furthermore, the residual solvent risks and the difficulty in completely removing silica traces pose additional regulatory hurdles for parenteral drugs.
The Novel Approach
The methodology disclosed in CN102001992A fundamentally reengineers the purification logic by exploiting the acid-base properties of the intermediate rather than relying solely on polarity differences. Instead of struggling to purify the free acid form of 1,4-dihydro-2,6-4-(2’,3’-dichlorophenyl)-5-carboxymethyl-3-pyridine carboxylic acid (Structure II), the process converts this intermediate directly into an alkali or alkaline earth metal salt. 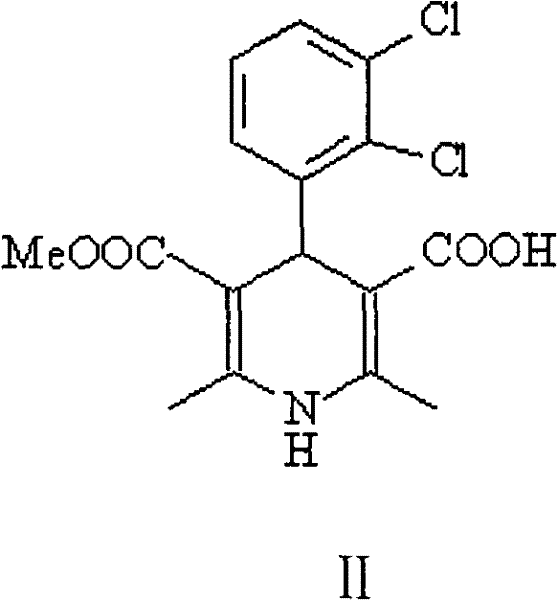 This ionic transformation dramatically alters the solubility profile of the molecule. While neutral impurities such as the symmetrical dimethyl diester remain soluble in the organic mother liquor or precipitate differently, the target salt can be selectively crystallized from aqueous or mixed solvent systems. This allows for the removal of the troublesome symmetric impurities without the need for chromatographic media. Once the high-purity salt is isolated, it serves as the direct substrate for the subsequent esterification, effectively decoupling the purification step from the final functionalization. This approach transforms a multi-step, solvent-intensive purification nightmare into a streamlined crystallization process that is inherently scalable and cost-effective.
This ionic transformation dramatically alters the solubility profile of the molecule. While neutral impurities such as the symmetrical dimethyl diester remain soluble in the organic mother liquor or precipitate differently, the target salt can be selectively crystallized from aqueous or mixed solvent systems. This allows for the removal of the troublesome symmetric impurities without the need for chromatographic media. Once the high-purity salt is isolated, it serves as the direct substrate for the subsequent esterification, effectively decoupling the purification step from the final functionalization. This approach transforms a multi-step, solvent-intensive purification nightmare into a streamlined crystallization process that is inherently scalable and cost-effective.
Mechanistic Insights into Salt-Mediated Purification and Esterification
The success of this synthetic route hinges on the precise control of ionic interactions and solubility equilibria during the salt formation phase. When the crude mono-acid intermediate is treated with a base such as sodium hydroxide, potassium hydroxide, or other alkali metal sources, the carboxylic acid moiety at the 3-position is deprotonated to form a carboxylate anion. This anionic species interacts strongly with the metal cation (Mn+), creating a lattice structure that is highly sensitive to solvent composition and temperature. The patent specifies that recrystallization can be effectively conducted in solvents ranging from C1-C4 alcohols to ketones and acetonitrile, often in mixture with water. The presence of water is particularly crucial as it enhances the solubility of the ionic salt at elevated temperatures while promoting precipitation upon cooling to ranges between -20°C and 35°C. This thermodynamic control ensures that the crystal lattice incorporates primarily the target salt, excluding neutral organic impurities that lack the charge necessary to integrate into the ionic crystal structure. The result is a purified salt intermediate with purity levels reaching 99.5% or higher, as confirmed by HPLC analysis in the patent examples.
Following purification, the mechanism shifts to a nucleophilic substitution reaction for the final esterification. Uniquely, the process does not require re-acidification of the salt to regenerate the free carboxylic acid. Instead, the carboxylate anion acts as a potent nucleophile, attacking the electrophilic carbon of chloromethyl n-butyrate in a polar aprotic solvent like DMF or acetonitrile. 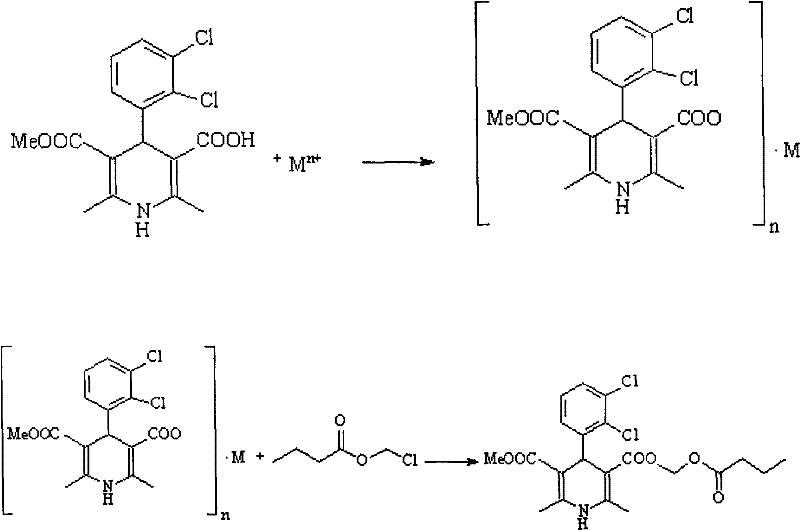 This direct displacement of the chloride ion by the carboxylate salt is highly efficient and minimizes side reactions. The use of the salt form actually enhances the reaction kinetics in polar solvents by ensuring the nucleophile is fully ionized and solvated. This mechanistic elegance eliminates an entire unit operation (acidification and extraction) from the process flow, reducing the overall processing time and minimizing the exposure of the sensitive dihydropyridine core to acidic conditions that could potentially lead to aromatization or degradation. The final product, clevidipine butyrate, is then isolated through standard workup and recrystallization, yielding a material with purity exceeding 99.9%, which is critical for injectable applications where particulate matter and impurities must be strictly controlled.
This direct displacement of the chloride ion by the carboxylate salt is highly efficient and minimizes side reactions. The use of the salt form actually enhances the reaction kinetics in polar solvents by ensuring the nucleophile is fully ionized and solvated. This mechanistic elegance eliminates an entire unit operation (acidification and extraction) from the process flow, reducing the overall processing time and minimizing the exposure of the sensitive dihydropyridine core to acidic conditions that could potentially lead to aromatization or degradation. The final product, clevidipine butyrate, is then isolated through standard workup and recrystallization, yielding a material with purity exceeding 99.9%, which is critical for injectable applications where particulate matter and impurities must be strictly controlled.
How to Synthesize Clevidipine Butyrate Efficiently
The implementation of this salt-mediated strategy offers a clear roadmap for process chemists aiming to optimize the production of clevidipine butyrate. The protocol begins with the preparation of the crude mono-acid intermediate, typically via a modified Hantzsch synthesis, followed immediately by salt formation without extensive intermediate purification. The choice of base and solvent system for recrystallization is flexible, allowing manufacturers to tailor the process to their existing infrastructure, whether utilizing sodium, potassium, or other compatible metal ions. The critical parameter is the temperature control during crystallization, which drives the exclusion of impurities. Once the pure salt is secured, the subsequent esterification proceeds smoothly under mild conditions, typically at room temperature or slightly below, ensuring the stability of the dihydropyridine ring. This sequence represents a significant departure from legacy methods, prioritizing operational simplicity and chemical efficiency.
- Convert the crude 1,4-dihydro-2,6-4-(2’,3’-dichlorophenyl)-5-carboxymethyl-3-pyridine carboxylic acid into an alkali or alkaline earth metal salt using bases like NaOH or KOH.
- Purify the resulting salt via recrystallization in water or organic solvent mixtures (e.g., ethanol/water) at temperatures between -20°C and 35°C to achieve >99.5% purity.
- React the purified salt directly with chloromethyl n-butyrate in DMF or acetonitrile to yield high-purity clevidipine butyrate without prior acidification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented technology translates into tangible operational improvements and risk mitigation. The most immediate impact is the drastic simplification of the manufacturing workflow. By eliminating the dependency on column chromatography, facilities can repurpose valuable production capacity that was previously tied up in slow, batch-wise purification cycles. This shift allows for a transition towards continuous or semi-continuous processing, which is the gold standard for modern chemical manufacturing. The removal of silica gel and the associated massive volumes of elution solvents also leads to substantial cost reduction in pharmaceutical manufacturing. Solvent recovery becomes more straightforward, and the waste disposal burden is significantly lightened, contributing to a greener and more sustainable production profile. These factors collectively enhance the economic viability of producing this high-value cardiovascular intermediate.
- Cost Reduction in Manufacturing: The elimination of column chromatography removes one of the most expensive unit operations in fine chemical synthesis. Silica gel is a consumable cost that scales linearly with production volume, and its disposal adds to environmental compliance costs. Furthermore, the solvents used in chromatography (often large volumes of hexanes, ethyl acetate, or dichloromethane) require significant energy for recovery or incineration. By replacing this with a crystallization-based purification, the process leverages the natural thermodynamic properties of the salt to achieve purity. This switch drastically reduces raw material consumption and utility costs associated with solvent handling. Additionally, the direct use of the salt in the esterification step saves the cost of acids and extra solvents that would otherwise be needed for acidification and extraction, compounding the savings throughout the synthesis.
- Enhanced Supply Chain Reliability: Supply chain resilience is often compromised by complex purification steps that are prone to bottlenecks. Chromatography columns can channel, clog, or fail to resolve impurities consistently, leading to batch failures and delayed shipments. The crystallization method described in this patent is far more robust and reproducible. Crystallization is a well-understood unit operation that scales predictably from the laboratory to the plant floor. This reliability ensures consistent lead times for high-purity pharmaceutical intermediates, allowing downstream API manufacturers to plan their production schedules with greater confidence. The ability to produce the intermediate in larger batch sizes without the throughput limitations of chromatography means that suppliers can respond more agilely to market demand surges for antihypertensive medications.
- Scalability and Environmental Compliance: As regulatory pressures regarding solvent emissions and waste generation intensify, processes that minimize environmental footprints are increasingly valued. This salt-formation route significantly reduces the E-factor (mass of waste per mass of product) of the synthesis. The solvents used for recrystallization, such as ethanol, methanol, or water mixtures, are generally less toxic and easier to manage than the halogenated solvents often required for chromatographic separations. The high purity achieved (>99.9%) also reduces the risk of downstream rejection by quality control labs, ensuring that every batch produced meets stringent specifications. This scalability makes the technology ideal for commercial scale-up of complex pharmaceutical intermediates, supporting the global demand for generic versions of clevidipine as patents on the originator drug expire.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. They are derived from the specific advantages and mechanistic details outlined in the patent documentation, providing clarity for technical teams evaluating this methodology for adoption. Understanding these nuances is essential for assessing the feasibility of integrating this process into existing manufacturing lines.
Q: Why is the salt formation step critical for purity in this synthesis?
A: The salt formation converts the target mono-acid intermediate into an ionic species, which exhibits significantly different solubility properties compared to neutral impurities like symmetrical diesters. This allows for effective removal of impurities via simple recrystallization, avoiding the need for complex chromatography.
Q: Can the purified salt be used directly for esterification?
A: Yes, a key innovation of this patent is that the purified salt does not require acidification back to the free acid form. It reacts directly with chloromethyl n-butyrate in polar aprotic solvents like DMF, streamlining the workflow and reducing waste.
Q: What are the scalability advantages of avoiding column chromatography?
A: Eliminating column chromatography removes a major bottleneck in industrial production. It drastically reduces solvent consumption, eliminates the cost of silica gel, and allows for continuous processing, making the method highly suitable for multi-ton commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Clevidipine Butyrate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and robust manufacturing capabilities. Our team has extensively analyzed the salt-formation strategy detailed in CN102001992A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We understand that achieving the stringent purity specifications required for injectable cardiovascular drugs demands more than just a recipe; it requires rigorous QC labs, state-of-the-art crystallization equipment, and a culture of quality that permeates every step of the process. Our facility is equipped to handle the specific solvent systems and temperature controls necessary to execute this high-purity synthesis efficiently, ensuring that the final clevidipine butyrate intermediate meets the highest global standards.
We invite pharmaceutical companies and contract manufacturers to collaborate with us to leverage this advanced technology for your supply chain. By partnering with us, you gain access to a Customized Cost-Saving Analysis that quantifies the specific economic benefits of switching to this chromatography-free route for your specific volume requirements. We encourage you to contact our technical procurement team today to request specific COA data from our pilot batches and to discuss route feasibility assessments tailored to your project timelines. Let us help you secure a stable, cost-effective, and high-quality supply of this critical antihypertensive intermediate.