Revolutionizing Carbetocin Manufacturing: Advanced All-Solid-Phase Synthesis and Commercial Scalability
Revolutionizing Carbetocin Manufacturing: Advanced All-Solid-Phase Synthesis and Commercial Scalability
The pharmaceutical landscape for peptide therapeutics is constantly evolving, driven by the need for higher purity, lower costs, and more sustainable manufacturing processes. A pivotal advancement in this domain is detailed in patent CN113801199A, which discloses a robust all-solid-phase synthesis method for Carbetocin, a long-acting oxytocin analogue critical for preventing postpartum hemorrhage. This technology represents a significant departure from legacy liquid-phase and semi-solid methods, offering a streamlined pathway that enhances both the chemical integrity of the final product and the economic efficiency of its production. By leveraging a unique combination of Fmoc solid-phase peptide synthesis (SPPS) and an on-resin Mitsunobu cyclization, this method addresses longstanding challenges in peptide manufacturing, such as low yields and difficult purification profiles. For global procurement leaders and R&D directors, understanding this technological shift is essential for securing reliable supply chains for high-value hormonal intermediates.
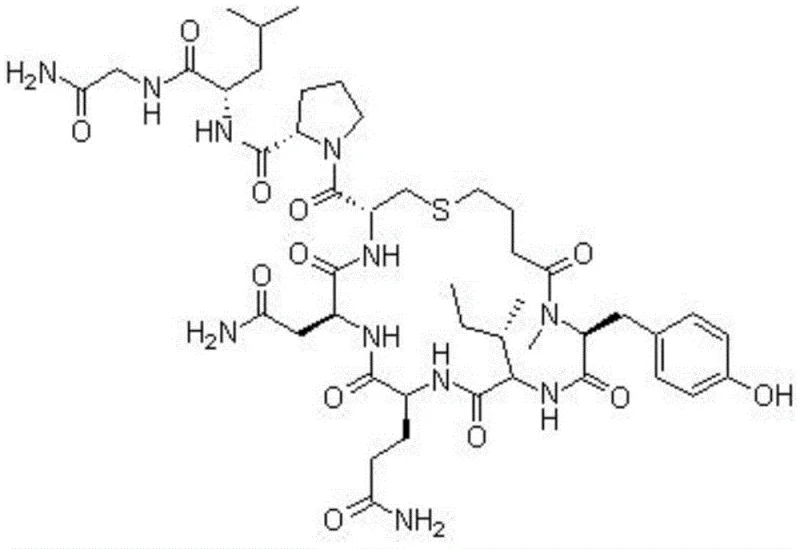
Carbetocin is a complex nonapeptide that requires precise stereochemical control and the formation of a specific thioether bridge to exhibit its biological activity. The molecular architecture, as illustrated in the structural diagram, demands a synthesis strategy that can handle multiple functional groups without cross-reactivity. Traditional approaches often struggled with the cyclization step, which is the most critical determinant of final purity. The innovation presented in CN113801199A replaces the conventional cysteine residue with serine and utilizes 4-mercaptobutyric acid, setting the stage for a highly selective intramolecular reaction. This strategic modification allows for the application of the Mitsunobu reaction directly on the solid support, a technique that offers superior control over reaction kinetics compared to solution-phase alternatives. For manufacturers of pharmaceutical intermediates, this translates to a process that is not only chemically elegant but also commercially viable for large-scale production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Carbetocin and its analogues has been plagued by inefficiencies inherent in liquid-phase and early-generation solid-phase techniques. Early liquid-phase synthesis processes were notoriously complex to operate, involving numerous isolation and purification steps that made them unsuitable for industrial-scale production. Even subsequent solid-liquid combined methods, such as those described in foreign patents ES2115543 and CS8605461, relied on liquid-phase cyclization. This approach necessitates reacting materials in extremely dilute solutions to favor intramolecular cyclization over intermolecular polymerization. The consequence is a massive consumption of organic solvents, complex post-treatment procedures, and the immediate generation of large volumes of hazardous waste liquid, creating significant environmental and cost burdens. Furthermore, domestic patents utilizing bromobutyric acid for cyclization faced issues with nucleophilic substitution; the active bromine atom could react indiscriminately with amino groups on the peptide chain rather than just the intended target. This lack of selectivity led to the formation of numerous impurities, lowering the overall yield and complicating the downstream purification process to meet stringent pharmacopeial standards.
The Novel Approach
The methodology outlined in CN113801199A fundamentally reengineers the cyclization step to overcome these historical bottlenecks. Instead of relying on halogenated hydrocarbons like bromobutyric acid or complex pre-coupled thioether strategies that require carboxyl protection, this invention employs a direct Mitsunobu reaction on the solid phase. By substituting cysteine with serine in the peptide sequence and introducing 4-mercaptobutyric acid at the N-terminus, the process creates a specific pairing between a hydroxyl group and a sulfhydryl group. Under the catalytic action of triphenylphosphine (PPh3) and diethyl azodicarboxylate (DEAD), these groups undergo dehydration to form the requisite thioether bond efficiently. This approach eliminates the need for the harsh conditions associated with alkali-mediated elimination reactions used in older methods. Moreover, it avoids the additional raw material costs and side reactions caused by decarboxylation protection found in other solid-phase strategies. The result is a synthesis route that significantly improves the purity of the crude peptide, reported to reach over 93% prior to final purification, thereby drastically reducing the load on preparative HPLC and increasing the overall recovery of the final API.
Mechanistic Insights into On-Resin Mitsunobu Cyclization
The core chemical innovation of this process lies in the execution of the Mitsunobu reaction within the constrained environment of a solid-phase resin matrix. In solution chemistry, Mitsunobu reactions are well-known for converting alcohols into various functional groups via an SN2 mechanism, but adapting this to solid-phase peptide synthesis (SPPS) requires precise control of steric and electronic factors. The mechanism begins with the activation of the serine side-chain hydroxyl group by the phosphonium intermediate formed from PPh3 and DEAD. This activation converts the poor leaving group (hydroxyl) into a highly reactive species. Subsequently, the terminal sulfhydryl group of the 4-mercaptobutyric acid moiety, which acts as a potent nucleophile, attacks the activated carbon center. Because both reactive groups are tethered to the same peptide chain anchored on the resin, the effective molarity for the intramolecular reaction is exceptionally high, favoring cyclization over intermolecular dimerization. This intramolecular preference is crucial for forming the cyclic structure of Carbetocin without generating linear polymers or oligomers. The mild conditions of the Mitsunobu reaction, typically conducted in tetrahydrofuran (THF) at room temperature, ensure that acid-labile side-chain protecting groups (like Trt) remaining on other residues (such as Asn and Gln) are not prematurely cleaved, maintaining the integrity of the peptide backbone during the critical ring-closing step.
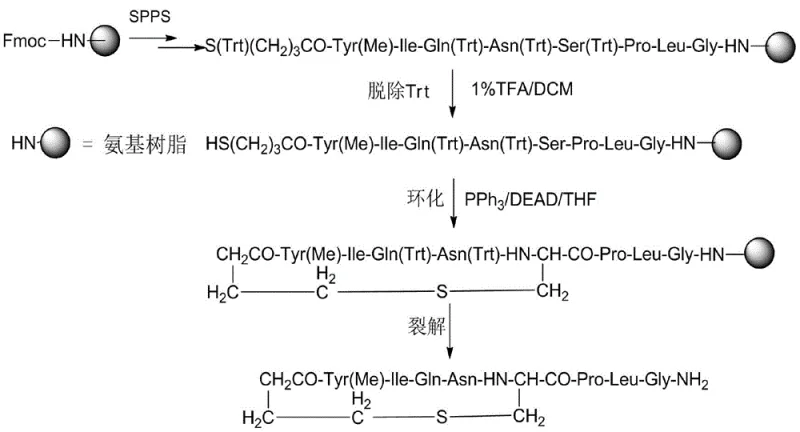
Impurity control is another critical aspect where this mechanistic approach excels. In traditional bromobutyric acid methods, the electrophilic bromine is susceptible to attack by any nucleophilic amine present in the peptide chain, leading to branched impurities that are structurally similar to the target and difficult to separate. In contrast, the Mitsunobu mechanism is highly chemoselective for the hydroxyl-thiol pair under the specified conditions. The use of dilute trifluoroacetic acid (1-5% TFA) for the selective deprotection of the Trt groups on the serine hydroxyl and the mercaptobutyric acid sulfur prior to cyclization ensures that only the intended reactive sites are exposed. This orthogonal deprotection strategy prevents premature side reactions. Furthermore, the avoidance of strong bases (like DIPEA in high concentrations for elimination reactions) reduces the risk of racemization at chiral centers, a common pitfall in peptide synthesis that can compromise the biological efficacy of the final drug. The comprehensive reaction scheme illustrates how the linear peptide resin is transformed into the cyclic product with minimal structural degradation, ensuring a clean impurity profile that facilitates easier downstream processing.
How to Synthesize Carbetocin Efficiently
The implementation of this all-solid-phase synthesis protocol requires strict adherence to the optimized parameters defined in the patent to achieve the reported high yields and purity. The process integrates standard Fmoc chemistry for chain elongation with specialized steps for cyclization and cleavage. Operators must carefully manage the stoichiometry of coupling reagents and the timing of deprotection events to prevent incomplete reactions or resin damage. The following overview outlines the critical operational phases, emphasizing the transition from linear assembly to cyclic formation. For detailed standard operating procedures and specific equipment requirements, please refer to the technical guidelines below.
- Sequentially couple protected amino acids (Gly, Leu, Pro, Ser, Asn, Gln, Ile, Tyr) and protected 4-mercaptobutyric acid onto Rink Amide resin to form linear peptide resin.
- Selectively remove the mercapto protecting group of 4-mercaptobutyric acid and the hydroxyl protecting group of serine using dilute TFA to expose reactive sites.
- Perform on-resin cyclization via Mitsunobu reaction using Triphenylphosphine (PPh3) and DEAD to form the critical thioether bond.
- Cleave the cyclic peptide from the resin using a TFA/TIS/Water cocktail and purify via reverse-phase HPLC to obtain high-purity Carbetocin.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route offers compelling economic and logistical benefits that extend beyond simple yield improvements. The shift to an all-solid-phase process with on-resin cyclization fundamentally alters the cost structure of Carbetocin manufacturing by eliminating several expensive and time-consuming unit operations. By avoiding the need for extremely dilute solutions required in liquid-phase cyclization, the process drastically reduces solvent consumption, which is a major cost driver in peptide synthesis. This reduction in solvent volume also translates to smaller reactor footprints and lower energy costs for solvent recovery and waste treatment. Additionally, the higher purity of the crude peptide means that the burden on the final purification step (typically preparative HPLC) is significantly reduced. Since purification is often the most costly and throughput-limiting step in peptide manufacturing, improving crude purity directly enhances production capacity and lowers the cost per gram of the final API. These efficiencies collectively contribute to a more resilient and cost-effective supply chain for this critical obstetric medication.
- Cost Reduction in Manufacturing: The elimination of bromobutyric acid and the associated complex protection/deprotection schemes simplifies the raw material bill of materials (BOM). By utilizing 4-mercaptobutyric acid directly and avoiding the side reactions caused by decarboxylation protection, the process reduces the consumption of expensive reagents and minimizes the loss of valuable peptide intermediates to side products. The qualitative improvement in reaction selectivity means less material is wasted, leading to substantial cost savings in raw material procurement. Furthermore, the simplified post-treatment workflow reduces labor hours and utility consumption, driving down the overall conversion cost.
- Enhanced Supply Chain Reliability: The robustness of the solid-phase method ensures consistent batch-to-batch quality, which is vital for regulatory compliance and supply continuity. Unlike liquid-phase methods that are sensitive to concentration fluctuations and mixing efficiency, the solid-phase approach provides a stable reaction environment that is easier to scale. The use of commercially available Fmoc-amino acids and standard coupling reagents ensures that the supply chain is not dependent on exotic or hard-to-source custom intermediates. This accessibility of raw materials mitigates the risk of supply disruptions and allows for flexible sourcing strategies, ensuring that production schedules can be maintained even in volatile market conditions.
- Scalability and Environmental Compliance: The process is inherently designed for scalability, moving seamlessly from laboratory benchtop to multi-kilogram production without the need for re-engineering the cyclization step. The reduction in solvent usage and the avoidance of hazardous halogenated byproducts align with modern green chemistry principles and increasingly stringent environmental regulations. This compliance reduces the regulatory burden on manufacturing sites and minimizes the risk of production stoppages due to environmental non-compliance. The ability to scale up while maintaining high purity standards makes this technology an ideal candidate for meeting the growing global demand for Carbetocin without compromising on sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the comparative data and experimental results presented in the patent documentation, providing a clear picture of the operational advantages. Understanding these nuances is critical for technical teams evaluating the feasibility of adopting this new manufacturing route.
Q: How does the Mitsunobu cyclization method improve Carbetocin purity compared to traditional methods?
A: Traditional methods using bromobutyric acid often suffer from nucleophilic substitution side reactions with amine groups, generating significant impurities. The novel Mitsunobu approach specifically targets the hydroxyl and sulfhydryl groups under mild conditions, drastically reducing side reactions and improving crude peptide purity from approximately 60% to over 93%.
Q: What are the supply chain advantages of this all-solid-phase synthesis route?
A: This method eliminates the need for complex liquid-phase cyclization steps which require extremely dilute solutions and vast amounts of solvent. By keeping the entire process on solid support, solvent consumption is reduced, post-treatment is simplified, and the process is inherently more scalable for industrial manufacturing, ensuring consistent supply continuity.
Q: Does this synthesis method avoid the use of expensive decarboxylation protection strategies?
A: Yes. Unlike previous solid-phase methods that required protecting the carboxyl group of butyric acid before coupling (which added cost and complexity), this invention utilizes 4-mercaptobutyric acid directly. This simplifies the raw material profile and removes side reactions associated with decarboxylation protection, leading to substantial cost optimization.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Carbetocin Supplier
As the global demand for high-quality peptide therapeutics continues to rise, partnering with a manufacturer that possesses advanced synthetic capabilities is paramount. NINGBO INNO PHARMCHEM stands at the forefront of this industry, leveraging cutting-edge technologies like the all-solid-phase Mitsunobu cyclization to deliver superior Carbetocin intermediates and APIs. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements without sacrificing quality. We operate with stringent purity specifications and utilize rigorous QC labs to guarantee that every batch meets the highest international standards, providing you with a reliable foundation for your pharmaceutical formulations.
We invite you to collaborate with us to optimize your supply chain and reduce your manufacturing costs. Our technical experts are ready to provide a Customized Cost-Saving Analysis tailored to your specific project needs, demonstrating how our advanced synthesis routes can improve your bottom line. Please contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you secure a sustainable and efficient supply of Carbetocin for your global markets.