Scalable Total Synthesis of Formalardeemin: A Strategic Breakthrough for Oncology API Manufacturing
Scalable Total Synthesis of Formalardeemin: A Strategic Breakthrough for Oncology API Manufacturing
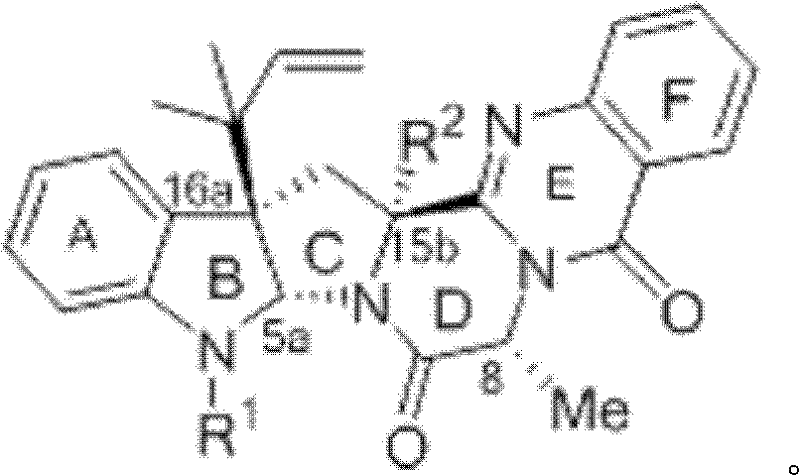
The escalating challenge of multidrug resistance (MDR) in oncology has intensified the global demand for potent chemosensitizers capable of reversing tumor cell tolerance to conventional chemotherapy. Patent CN102276615A, filed in December 2011, presents a pivotal advancement in the medicinal chemistry landscape by disclosing a highly efficient total synthesis method for (-)-5-N-formalardeemin, a derivative of the fungal metabolite ardeemin. This compound has demonstrated superior efficacy in reversing MDR compared to its acetyl counterpart, making it a critical candidate for next-generation cancer therapies. The disclosed methodology addresses the historical bottleneck of supply scarcity, transitioning the production paradigm from unreliable natural extraction to a robust, reproducible chemical synthesis. For pharmaceutical developers and supply chain strategists, this patent represents a viable pathway to secure high-purity intermediates essential for preclinical and clinical development programs.
From a strategic sourcing perspective, the ability to synthesize complex alkaloids like formalardeemin through a defined chemical route rather than relying on biological fermentation fundamentally alters the risk profile of the supply chain. Natural extraction is often plagued by low titers, complex isolation procedures, and batch-to-batch variability inherent in biological systems. In contrast, the synthetic approach detailed in this patent utilizes standard organic transformations that can be rigorously controlled and optimized. This shift not only guarantees a consistent supply of the active pharmaceutical ingredient (API) intermediate but also opens the door for structural analoging, allowing R&D teams to explore structure-activity relationships (SAR) more freely. As a reliable pharmaceutical intermediate supplier, understanding these synthetic nuances is key to delivering value to partners seeking to mitigate supply risks in their oncology pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the acquisition of ardeemin and its bioactive derivatives relied heavily on the extraction from fungal cultures such as Aspergillus fischerii. This conventional approach is fraught with significant logistical and economic inefficiencies that hinder large-scale drug development. The fermentation process yields extremely low concentrations of the target metabolites, necessitating the processing of vast volumes of culture broth to isolate minute quantities of the active compound. Furthermore, the downstream purification from a complex biological matrix involves tedious chromatographic separations to remove structurally similar impurities and fungal byproducts, driving up the cost of goods sold (COGS) prohibitively. Beyond economic factors, the reliance on biological sources introduces unacceptable volatility into the supply chain; factors such as strain degeneration, contamination, and seasonal variations in fermentation efficiency can lead to sudden supply disruptions. For procurement managers overseeing oncology projects, depending on such an unstable source for a critical MDR reversal agent poses a substantial threat to project timelines and regulatory filings.
The Novel Approach
The synthetic strategy outlined in patent CN102276615A offers a transformative solution by establishing a concise, eight-step total synthesis route that bypasses the limitations of natural extraction entirely. This novel approach leverages a convergent synthesis design, starting from readily available indoline precursors and constructing the complex pentacyclic core through a series of high-yielding transformations. A key highlight of this methodology is the initial silver-mediated allylation step, which efficiently installs the crucial prenyl side chain with high stereocontrol, setting the stage for the subsequent ring closures. By replacing the unpredictable biology of fermentation with the precision of organic synthesis, this method achieves a total yield exceeding 30%, a figure that is remarkably high for molecules of this structural complexity. This efficiency translates directly into reduced raw material consumption and waste generation, aligning with modern green chemistry principles while ensuring economic viability for commercial scale-up of complex alkaloids.
Mechanistic Insights into Silver-Catalyzed Allylation and Cyclization
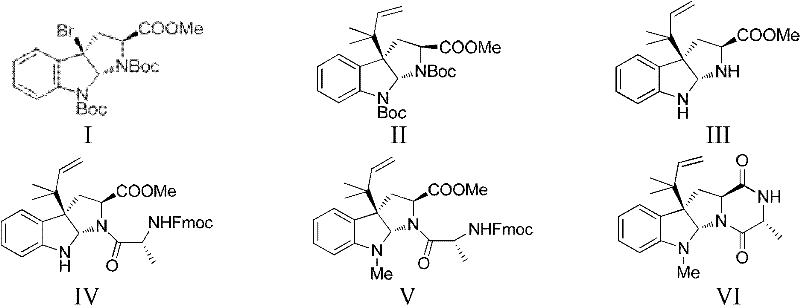
The cornerstone of this synthetic route lies in the sophisticated application of organometallic chemistry to construct the sterically crowded quaternary center at the C3 position of the indoline ring. In the first critical step, the reaction between the bromo-indoline precursor and tri-n-butyl(3-methyl-2-butenyl)tin is facilitated by a strong base and a silver salt, such as silver perchlorate. Mechanistically, the silver ion acts as a halophilic Lewis acid, promoting the ionization of the carbon-bromine bond to generate a reactive cationic intermediate at the C3 position. This electrophilic species is then trapped by the nucleophilic allylstannane reagent in a stereoselective manner, driven by the chiral environment of the existing scaffold. This transformation is particularly noteworthy because it avoids the use of expensive palladium catalysts often required for similar cross-coupling reactions, thereby offering a cost reduction in API manufacturing by utilizing more abundant silver salts. The precise control of reaction temperature, typically maintained at cryogenic conditions like -78°C, is essential to suppress side reactions and ensure the formation of the desired diastereomer, which is critical for the biological activity of the final product.
Following the construction of the linear precursor, the synthesis proceeds through a cascade of cyclization and functionalization events that demonstrate excellent chemoselectivity. The formation of the quinazolinone ring system, a defining feature of the ardeemin skeleton, is achieved through the reaction with o-azidobenzoic anhydride followed by a Staudinger-type reduction and intramolecular cyclization. This sequence elegantly installs the nitrogen-containing heterocycle while simultaneously reducing the azide functionality, minimizing the number of isolation steps required. The final oxidative conversion of the N-methyl group to the N-formyl group is executed using mild oxidizing agents like pyridinium chlorochromate (PCC) or activated manganese dioxide. This late-stage oxidation is highly selective, targeting the indole nitrogen without affecting other sensitive functionalities such as the olefinic bonds or the amide linkages within the molecule. Such mechanistic precision ensures a clean impurity profile, simplifying the purification burden and facilitating the production of high-purity MDR reversal agents suitable for rigorous pharmacodynamic evaluation.
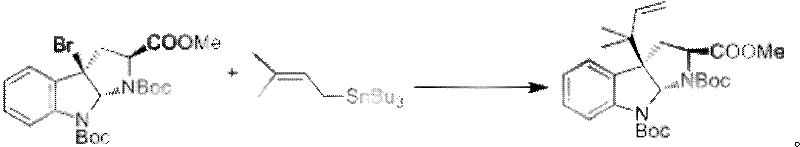
How to Synthesize (-)-5-N-formalardeemin Efficiently
The execution of this synthesis requires strict adherence to the optimized reaction conditions detailed in the patent to maximize yield and purity. The process begins with the preparation of the key allylated intermediate under inert atmosphere to prevent moisture sensitivity, followed by a carefully orchestrated sequence of deprotection and coupling reactions. Each step has been validated to proceed with high conversion rates, minimizing the accumulation of intermediates that could complicate downstream processing. For process chemists looking to implement this route, it is imperative to focus on the purification strategies employed after the cyclization steps, as these are critical junctures for removing regioisomers. The detailed standardized synthesis steps see the guide below for a comprehensive breakdown of the operational parameters.
- Perform silver-mediated allylation of the bromo-indoline precursor using tri-n-butyl(3-methyl-2-butenyl)tin to establish the core carbon skeleton.
- Execute sequential deprotection, amide coupling, and indole N-methylation to functionalize the intermediate scaffold.
- Complete the synthesis via cyclization with o-azidobenzoic anhydride, azide reduction, and final oxidative formylation to yield the target molecule.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers compelling advantages that extend beyond mere technical feasibility. The primary value proposition lies in the decoupling of supply from biological constraints, ensuring a predictable and continuous flow of materials essential for clinical trials and eventual commercial launch. By utilizing commodity chemicals and standard reactor setups, the barrier to entry for manufacturing is significantly lowered, allowing for flexible production scaling based on market demand. This flexibility is crucial in the volatile landscape of oncology drug development, where project priorities can shift rapidly. Furthermore, the robustness of the synthetic steps reduces the risk of batch failures, a common issue in complex natural product synthesis, thereby enhancing overall supply chain reliability and reducing the need for safety stock holdings.
- Cost Reduction in Manufacturing: The elimination of fermentation and extraction processes removes the need for specialized bioreactors and extensive downstream purification infrastructure, leading to substantial capital expenditure savings. Additionally, the use of inexpensive reagents such as cesium carbonate and formaldehyde, instead of costly enzymes or rare earth catalysts, drives down the variable cost per kilogram. The high overall yield of the process means that less raw material is wasted, further optimizing the cost structure and making the final API more economically attractive for inclusion in combination therapy regimens.
- Enhanced Supply Chain Reliability: Synthetic routes are inherently more stable than biological sources, as they are not subject to the vagaries of microbial strain performance or environmental factors. This stability allows for long-term supply agreements with fixed quality specifications, providing peace of mind to R&D teams planning multi-year development programs. The ability to produce the intermediate on demand reduces lead times significantly, enabling faster response to clinical data updates and regulatory requirements. Moreover, the modular nature of the synthesis allows for parallel processing of different intermediates, further compressing the overall production timeline.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing solvents and reaction conditions that are compatible with standard glass-lined steel reactors used in the fine chemical industry. This compatibility facilitates a smooth technology transfer from laboratory to pilot plant and eventually to full commercial production without the need for major process re-engineering. From an environmental perspective, the shorter reaction sequence and higher atom economy result in a reduced E-factor (mass of waste per mass of product), aligning with increasingly stringent global environmental regulations and corporate sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of formalardeemin intermediates. These insights are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for industry stakeholders. Understanding these details is crucial for making informed decisions about sourcing and development strategies.
Q: What is the primary advantage of this synthetic route over fungal extraction?
A: Unlike fungal extraction which suffers from low yields and seasonal variability, this total synthesis offers a consistent, scalable supply chain with a total yield exceeding 30%, ensuring reliable availability for clinical research.
Q: How does the process ensure high purity for pharmacodynamic studies?
A: The route utilizes robust purification techniques such as silica gel column chromatography at critical intermediate stages, effectively removing side products and ensuring the stringent purity specifications required for biological testing.
Q: Is this methodology suitable for industrial scale-up?
A: Yes, the protocol employs commercially available reagents like cesium carbonate and standard solvents like dichloromethane, avoiding exotic catalysts, which significantly simplifies the transition from laboratory gram-scale to multi-kilogram commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Formalardeemin Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of life-saving oncology therapies. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and reliability. We are committed to delivering compounds that meet stringent purity specifications through our rigorous QC labs, which utilize state-of-the-art analytical instrumentation to verify identity and assay. By partnering with us, you gain access to a supply chain that is both resilient and responsive, capable of adapting to the dynamic requirements of the pharmaceutical industry.
We invite you to contact our technical procurement team to discuss how we can support your specific project requirements. Whether you need a Customized Cost-Saving Analysis for your current supply chain or require specific COA data to validate our quality standards, we are here to assist. Let us provide you with detailed route feasibility assessments to help you navigate the complexities of bringing new MDR reversal agents to market. Reach out today to secure a reliable supply of formalardeemin and accelerate your drug development timeline.