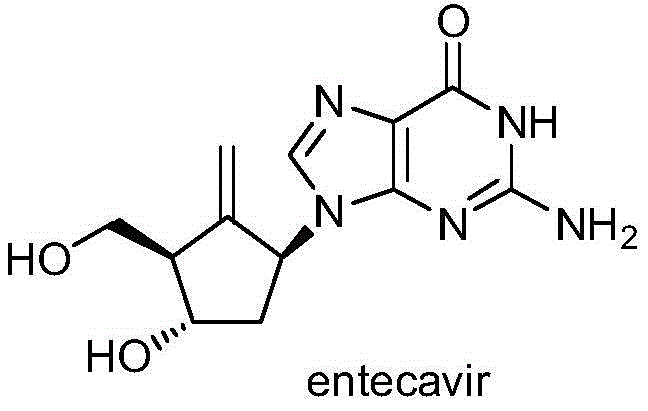
Advanced Entecavir Synthesis: High-Yield Intermediates for Commercial API Production
The pharmaceutical industry continuously seeks more efficient pathways for producing critical antiviral agents, and the recent disclosure in patent CN112625041A represents a significant leap forward in the manufacturing of Entecavir. This potent deoxyguanosine analogue, widely recognized for its efficacy against the hepatitis B virus, has traditionally been synthesized through routes plagued by low yields and complex purification requirements. The new methodology introduces novel intermediates, specifically compounds of Formula II and Formula III, which fundamentally alter the reaction landscape. By implementing a strategic protection-deprotection sequence on the purine base, the inventors have achieved a synthesis that not only enhances the overall yield but also drastically simplifies the downstream processing. For R&D directors and procurement specialists alike, this patent offers a compelling blueprint for reducing the cost of goods sold (COGS) while maintaining the stringent quality standards required for antiviral APIs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Entecavir has relied on routes that introduce chiral centers gradually, often starting from simple precursors like cyclopentadiene. As illustrated in earlier patents such as US5206244, these methods suffer from significant drawbacks, primarily the difficulty in controlling optical impurities. The gradual construction of the stereochemical framework necessitates multiple resolution steps or expensive chiral catalysts, which inherently limits the overall yield. Furthermore, alternative routes utilizing (+)-Corylactone diol, while solving the chirality issue, have encountered bottlenecks in the coupling stage. The direct reaction between the cyclopentyl intermediate and unprotected 2-amino-6-chloropurine often results in poor conversion rates and the formation of difficult-to-remove byproducts. This inefficiency forces manufacturers to rely on resource-intensive column chromatography for purification, a practice that is economically unsustainable and environmentally burdensome at a commercial scale.
The Novel Approach
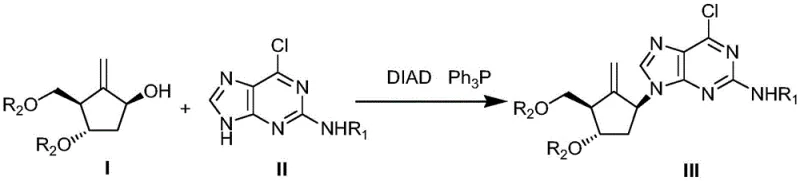
The innovative strategy detailed in CN112625041A circumvents these historical bottlenecks by pre-modifying the purine base before the coupling event. Instead of reacting the naked amine, the process employs a protected intermediate (Formula II) where the exocyclic amino group is masked with bulky groups such as trityl or (4-methoxyphenyl)benzhydryl. This modification serves a dual purpose: it enhances the solubility of the purine derivative in organic solvents and, more critically, prevents unwanted side reactions at the amino position during the subsequent coupling. The result is a highly efficient Mitsunobu reaction that proceeds with exceptional cleanliness. By shifting the complexity to the preparation of a stable intermediate, the final coupling step becomes robust and high-yielding, effectively eliminating the need for preparative HPLC or silica gel columns in the final stages of API production.
Mechanistic Insights into Mitsunobu Coupling with Protected Purines
The core of this technological advancement lies in the meticulous application of the Mitsunobu reaction, a staple in nucleoside chemistry, but optimized here through electronic and steric tuning of the nucleophile. In the standard Mitsunobu protocol, an alcohol is activated by a phosphine and an azodicarboxylate (such as DIAD) to form an alkoxyphosphonium salt, which is then displaced by a nucleophile. In the context of Entecavir synthesis, the nucleophile is the N9 nitrogen of the purine ring. However, the presence of a free exocyclic amine at the C2 position in traditional substrates often leads to competitive N-alkylation or polymerization, degrading the yield. The patent solves this by converting 2-amino-6-chloropurine into Formula II-1 using di-tert-butyl dicarbonate, followed by alkylation with R1Cl to generate Formula II. This N-protection effectively deactivates the exocyclic amine, directing the nucleophilic attack exclusively to the N9 position. The steric bulk of the trityl or MMB group further shields the molecule, ensuring that the inversion of configuration at the cyclopentyl center proceeds with high fidelity, preserving the critical (1S,3R,4S) stereochemistry required for biological activity.
Furthermore, the choice of protecting groups is not arbitrary but is engineered for facile removal in the final step. The patent demonstrates that both trityl and (4-methoxyphenyl)benzhydryl groups are acid-labile, allowing for a unified deprotection strategy. After the coupling yields the protected nucleoside (Formula III), the final transformation involves a simple acid hydrolysis using hydrochloric acid in methanol. This single operation simultaneously cleaves the silyl or benzyl ethers on the cyclopentyl ring and removes the bulky amine protectors on the purine base. The mechanistic elegance here is that the harsh conditions required to remove the trityl group also facilitate the hydrolysis of the silyl ethers, converging multiple synthetic operations into one pot. This convergence minimizes the number of isolation steps, thereby reducing material loss and solvent consumption, which are key drivers in the economic viability of pharmaceutical manufacturing.
How to Synthesize Entecavir Efficiently
The synthesis of Entecavir via this novel route is designed for operational simplicity and high throughput, making it an ideal candidate for technology transfer from the laboratory to the pilot plant. The process begins with the preparation of the protected purine intermediate, followed by the critical coupling with the chiral cyclopentyl building block, and concludes with a global deprotection. Each step has been optimized to maximize yield and minimize impurity profiles, ensuring that the final API meets regulatory specifications without extensive rework. The following guide outlines the standardized synthetic steps derived from the patent examples, providing a clear roadmap for process chemists aiming to implement this superior methodology.
- Protect 2-amino-6-chloropurine using di-tert-butyl dicarbonate followed by trityl or MMB chloride to form the stable Formula II intermediate.
- Perform a Mitsunobu coupling reaction between the protected purine (Formula II) and the cyclopentyl alcohol (Formula I) using DIAD and triphenylphosphine.
- Execute global deprotection and hydrolysis using hydrochloric acid in methanol to yield high-purity Entecavir with minimal optical impurities.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route translates directly into enhanced supply security and reduced manufacturing costs. The traditional reliance on column chromatography for purification is a major vulnerability in the supply chain, as it limits batch sizes and increases cycle times. By replacing this with crystallization and extraction, the new method significantly increases the throughput capacity of existing manufacturing facilities. Moreover, the high yields reported in the patent examples—reaching up to 98% for the intermediate coupling and 68% for the final API step—imply a substantial reduction in the consumption of raw materials. This efficiency gain is critical in a market where the demand for antiviral medications remains volatile and high.
- Cost Reduction in Manufacturing: The elimination of expensive chromatographic purification steps is the primary driver for cost savings in this process. In conventional synthesis, the loss of material during column separation can be significant, often exceeding 20-30% of the batch. By achieving high purity through crystallization, as demonstrated by the 99.8% HPLC purity in the patent examples, manufacturers can recover nearly all theoretical yield. Additionally, the use of commodity reagents like triphenylphosphine and DIAD, which are readily available in bulk, ensures that the cost of goods remains stable and predictable, shielding the supply chain from the volatility associated with specialized chiral catalysts.
- Enhanced Supply Chain Reliability: The robustness of the Mitsunobu coupling under the described conditions (-40°C to room temperature) allows for flexible scheduling and easier scale-up. Unlike enzymatic resolutions or sensitive organometallic reactions that require strict anhydrous conditions and inert atmospheres throughout, this chemical synthesis is tolerant of minor variations, reducing the risk of batch failures. The availability of the starting materials, particularly 2-amino-6-chloropurine and the cyclopentyl diol derivatives, from multiple global suppliers further mitigates the risk of single-source dependency, ensuring a continuous flow of intermediates for API production.
- Scalability and Environmental Compliance: From an environmental perspective, the reduction in solvent usage is a significant advantage. Chromatography typically consumes vast quantities of organic solvents, creating a heavy burden on waste treatment facilities. By shifting to a crystallization-based purification, the volume of hazardous waste generated per kilogram of API is drastically reduced. This aligns with modern green chemistry principles and simplifies regulatory compliance regarding solvent emissions and waste disposal. The ability to run the final deprotection in a methanol/water system also facilitates easier solvent recovery and recycling, contributing to a more sustainable and cost-effective manufacturing lifecycle.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel Entecavir synthesis route. These answers are derived directly from the experimental data and claims presented in patent CN112625041A, providing factual insights for decision-makers evaluating this technology for their production pipelines.
Q: What is the primary advantage of the new intermediate Formula II in Entecavir synthesis?
A: The primary advantage is the significant improvement in coupling yield and purity. By protecting the exocyclic amine of 2-amino-6-chloropurine (using Trityl or MMB groups), side reactions during the Mitsunobu coupling are suppressed, leading to yields as high as 98% for the coupled intermediate compared to lower yields in conventional routes.
Q: How does this method address the optical purity challenges of previous Entecavir routes?
A: This method utilizes a chiral cyclopentyl starting material (Formula I) where the stereocenters are already established. The mild Mitsunobu conditions and the specific protecting groups prevent racemization, ensuring the final API achieves an HPLC purity of 99.8% without the need for extensive column chromatography.
Q: Is this synthetic route suitable for large-scale commercial manufacturing?
A: Yes, the route is highly scalable. It replaces difficult purification steps like column chromatography with standard crystallization and extraction techniques. The use of robust reagents like DIAD and triphenylphosphine in common solvents like THF allows for straightforward scale-up from kilogram to multi-ton production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Entecavir Supplier
At NINGBO INNO PHARMCHEM, we understand that the transition from a patented laboratory method to a commercial reality requires deep technical expertise and rigorous quality control. Our team specializes in scaling complex pharmaceutical pathways, leveraging our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We are equipped with state-of-the-art reactors capable of handling the low-temperature requirements of the Mitsunobu reaction and the corrosive conditions of the final hydrolysis step. Our commitment to quality is unwavering, with stringent purity specifications enforced through our rigorous QC labs, ensuring that every batch of Entecavir intermediate or API we produce meets the highest international pharmacopeial standards.
We invite pharmaceutical partners to collaborate with us to unlock the full potential of this cost-effective synthesis route. Whether you require custom synthesis of the protected intermediates or full-scale API manufacturing, our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our advanced manufacturing capabilities can optimize your Entecavir supply chain.