Advanced Synthesis of Antiviral Intermediate BL: Technical Breakthroughs and Commercial Scalability
Advanced Synthesis of Antiviral Intermediate BL: Technical Breakthroughs and Commercial Scalability
The global pharmaceutical landscape is continuously evolving, driven by the urgent need for effective antiviral therapeutics. In this context, Patent CN115894498A introduces a significant advancement in the synthesis of Intermediate BL, a critical precursor for potential new antiviral compounds analogous to Remdesivir. This patent details a robust three-step synthetic route that addresses key challenges in stereocontrol and functional group modification on the pyrrolo[2,1-f][1,2,4]triazine core. Unlike previous methods that left the 4-amino group unmodified, this innovation allows for precise acylation, opening doors to novel drug candidates with improved pharmacokinetic profiles. For R&D directors and procurement specialists, understanding this pathway is essential for securing a reliable supply of high-quality nucleoside analogs.
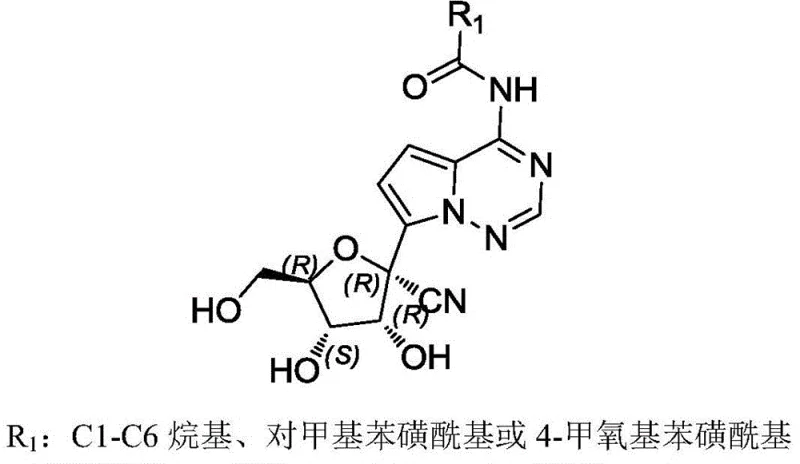
The core value of this technology lies in its ability to produce Intermediate BL with exceptional purity and chiral integrity. The structure features a complex ribose-like sugar moiety fused to a heterocyclic base, with specific stereochemical configurations at multiple centers denoted as (R) and (S). The versatility of the R1 group, which can be a C1-C6 alkyl, p-toluenesulfonyl, or 4-methoxybenzenesulfonyl moiety, provides medicinal chemists with a powerful handle for structure-activity relationship (SAR) studies. As a reliable pharmaceutical intermediate supplier, recognizing the strategic importance of such versatile scaffolds is key to supporting downstream drug development pipelines effectively.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of nucleoside analogs similar to Remdesivir has been plagued by issues regarding regioselectivity and the difficulty of modifying the exocyclic amine without affecting other sensitive functional groups. Traditional routes often suffered from poor diastereoselectivity during the formation of the glycosidic bond or the introduction of the nitrile group, leading to complex mixtures that were difficult and costly to separate. Furthermore, many existing processes relied on harsh conditions or expensive transition metal catalysts that posed significant challenges for waste management and regulatory compliance in GMP environments. These inefficiencies resulted in prolonged lead times and inflated costs, creating bottlenecks for the commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
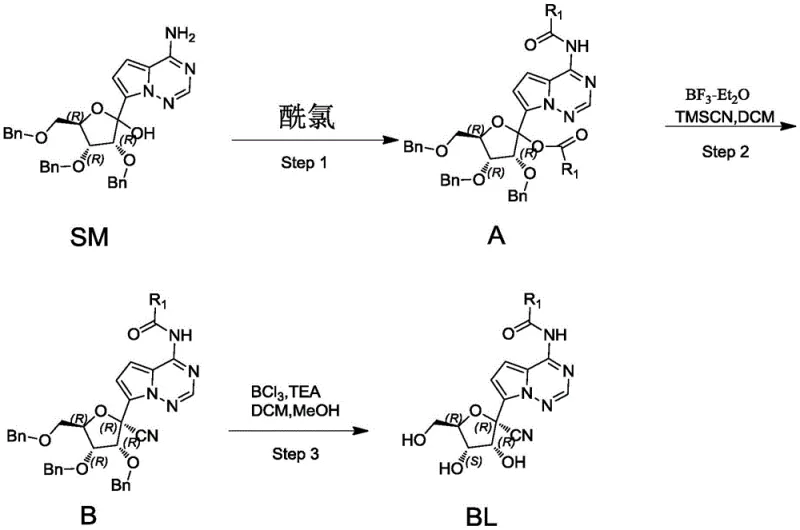
The methodology described in Patent CN115894498A offers a transformative solution by streamlining the synthesis into three highly efficient steps. The process begins with the selective acylation of the starting material (SM) to protect the amino and hydroxyl groups simultaneously, forming Intermediate A. This is followed by a Lewis acid-catalyzed cyanation using TMSCN, which proceeds with remarkable diastereoselectivity to yield Intermediate B. Finally, a mild debenzylation step unveils the free hydroxyl groups to afford the target Intermediate BL. This route eliminates the need for cumbersome purification steps and avoids the use of heavy metals, representing a paradigm shift towards greener and more cost-effective manufacturing.
Mechanistic Insights into Lewis Acid-Catalyzed Stereoselective Cyanation
The heart of this synthetic innovation is the second step, where Intermediate A undergoes cyanation in the presence of boron trifluoride etherate (BF3-Et2O) and trimethylsilyl cyanide (TMSCN). Mechanistically, the Lewis acid activates the hemiacetal or oxocarbenium ion intermediate generated from the protected sugar moiety. This activation facilitates the nucleophilic attack by the cyanide ion from TMSCN. Crucially, the steric environment created by the adjacent protecting groups and the fused ring system directs the attack to occur predominantly from one face, resulting in the high diastereoselectivity observed. This control is vital for ensuring that the final product possesses the correct biological configuration, thereby maximizing potency and minimizing off-target effects.
Furthermore, the choice of the R1 group plays a subtle yet significant role in stabilizing the transition state and influencing the electronic properties of the triazine ring. Whether utilizing a bulky pivaloyl group or an electron-withdrawing sulfonyl group, the process maintains high fidelity. For instance, when R1 is a pivaloyl group, the steric bulk helps shield certain reactive sites, preventing side reactions. This level of mechanistic understanding allows process chemists to fine-tune reaction conditions, such as temperature and stoichiometry, to optimize yields further. Such precision is what distinguishes a laboratory curiosity from a commercially viable process capable of meeting stringent quality specifications.
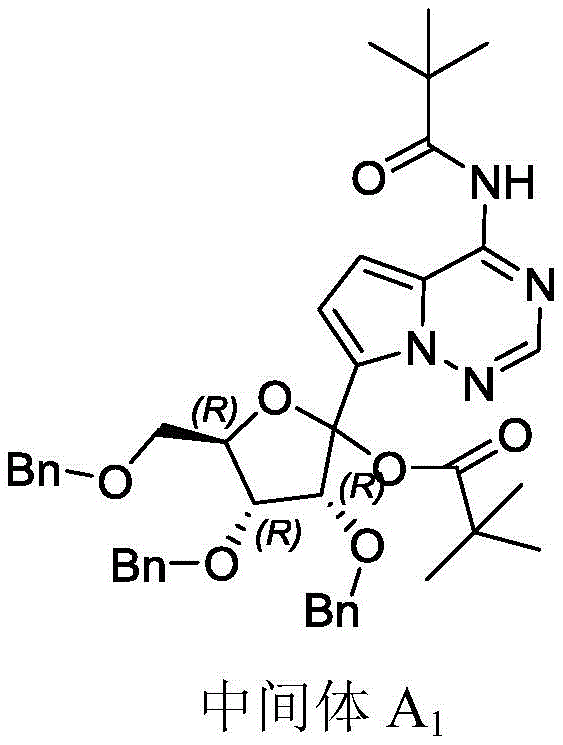
How to Synthesize Intermediate BL Efficiently
Executing this synthesis requires careful attention to temperature control and reagent addition rates to maintain safety and reproducibility. The process is designed to be scalable, utilizing common solvents like dichloromethane (DCM) and reagents that are readily available in the global chemical market. The initial acylation is performed under nitrogen protection to prevent moisture interference, followed by a controlled workup to isolate Intermediate A. The subsequent cyanation step demands low-temperature initiation to manage exotherms before warming to promote conversion. Finally, the debenzylation utilizes boron trichloride, a potent reagent that requires quenching with methanol to safely release the final product. Detailed standardized operating procedures are essential for transferring this technology from pilot plants to full-scale production facilities.
- Step 1: Acylation of SM with acyl chloride using TEA and DMAP in DCM at controlled temperatures to form Intermediate A.
- Step 2: Reaction of Intermediate A with TMSCN catalyzed by BF3-Et2O to achieve high diastereoselectivity, yielding Intermediate B.
- Step 3: Debenzylation of Intermediate B using BCl3 in DCM at low temperatures to afford the final Intermediate BL.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible benefits that extend beyond mere technical feasibility. The simplicity of the operation translates directly into reduced operational expenditures (OPEX) and lower capital investment requirements for equipment. By avoiding exotic catalysts and complex multi-step sequences, manufacturers can significantly reduce the risk of batch failures and supply disruptions. Moreover, the high purity of the crude product minimizes the need for extensive chromatographic purification, which is often a major cost driver in pharmaceutical manufacturing. This efficiency ensures a more stable and predictable supply of critical intermediates for downstream API production.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts removes the necessity for expensive metal scavenging steps and rigorous residual metal testing, which are costly and time-consuming. Additionally, the high yields reported in the patent examples suggest that raw material consumption is optimized, leading to substantial cost savings per kilogram of product. The use of commodity chemicals like TMSCN and acyl chlorides further insulates the supply chain from volatility associated with specialized reagents. These factors combined create a leaner manufacturing model that enhances overall profitability.
- Enhanced Supply Chain Reliability: The robustness of this three-step sequence means that production timelines can be shortened significantly compared to legacy routes. The reagents involved are widely sourced from multiple global suppliers, reducing the risk of single-source dependency. Furthermore, the process tolerates a range of R1 groups, providing flexibility to switch protecting groups based on availability without redesigning the entire synthetic pathway. This adaptability is crucial for maintaining continuity of supply in a dynamic global market where logistical challenges are frequent.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, with reaction conditions that are easily manageable in large reactors. The avoidance of heavy metals aligns with increasingly strict environmental regulations and corporate sustainability goals. Waste streams are simpler to treat, primarily consisting of organic solvents and salts that can be recovered or disposed of using standard protocols. This environmental compatibility not only reduces disposal costs but also enhances the company's reputation as a responsible manufacturer, which is increasingly important for partnerships with top-tier pharmaceutical companies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis of Intermediate BL. These answers are derived directly from the technical specifications and experimental data provided in the patent documentation. They serve to clarify the capabilities of this method and its suitability for various applications in antiviral drug development. Understanding these details helps stakeholders make informed decisions about integrating this technology into their existing portfolios.
Q: What is the key advantage of the cyanation step in this synthesis?
A: The use of Lewis acid BF3-Et2O with TMSCN ensures very high diastereoselectivity at the anomeric center, crucial for biological activity.
Q: Can the R1 group be varied in this process?
A: Yes, the process supports R1 groups including C1-C6 alkyl, p-toluenesulfonyl, or 4-methoxybenzenesulfonyl, allowing for diverse derivative synthesis.
Q: Is this process suitable for large-scale manufacturing?
A: Absolutely. The patent highlights simple operations, high yields, and the use of standard reagents, making it highly suitable for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Intermediate BL Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the race to develop new antiviral therapies. Our team of expert chemists has extensively analyzed the route described in Patent CN115894498A and is fully prepared to execute this synthesis with precision. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met regardless of the stage of development. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of Intermediate BL meets the highest industry standards for chirality and chemical purity.
We invite you to collaborate with us to leverage this advanced synthetic technology for your next-generation antiviral programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact us today to request specific COA data and route feasibility assessments. By partnering with NINGBO INNO PHARMCHEM, you gain access to a supply chain partner committed to innovation, quality, and reliability in the field of pharmaceutical intermediates.