Advanced Synthesis of Tenofovir Disoproxil Fumarate for Commercial Scale-up
Introduction to Novel Tenofovir Disoproxil Fumarate Synthesis
The global demand for effective antiretroviral therapies continues to drive innovation in the manufacturing of key active pharmaceutical ingredients (APIs), particularly Tenofovir Disoproxil Fumarate (TDF), a cornerstone treatment for HIV-1 and Hepatitis B. Patent CN103360425A introduces a transformative synthesis method that addresses critical bottlenecks in the conventional production of this vital medication. Unlike traditional routes that rely on cumbersome azeotropic dehydration and high-boiling solvents, this invention leverages a direct condensation strategy utilizing solid dehydration agents and optimized solvent systems. This technical breakthrough not only enhances the chemical purity and molar yield of the final product but also aligns with modern green chemistry principles by minimizing the use of toxic volatile organic compounds. For pharmaceutical manufacturers and procurement specialists, understanding this shift is essential for securing a reliable API intermediate supplier capable of delivering high-quality materials with improved cost-efficiency and supply chain resilience.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historical manufacturing processes for Tenofovir Disoproxil have been plagued by significant operational inefficiencies and environmental concerns that hinder cost reduction in pharmaceutical manufacturing. Traditional protocols typically necessitate an azeotropic distillation step using solvents such as cyclohexane, n-hexane, or toluene to remove crystal water from Tenofovir monohydrate prior to the condensation reaction. This dehydration process is notoriously time-consuming, often requiring reflux periods exceeding 24 hours, which drastically reduces reactor throughput and increases energy consumption. Furthermore, the transfer of dehydrated materials poses a high risk of moisture re-absorption from the atmosphere, which can severely compromise the subsequent condensation yield. Additionally, the widespread use of N-Methyl pyrrolidone (NMP) as a reaction solvent creates severe downstream processing challenges; its high boiling point and viscosity make it extremely difficult to remove completely, leading to thick reaction mixtures that complicate suction filtration and often result in residual solvent levels exceeding strict regulatory standards.
The Novel Approach
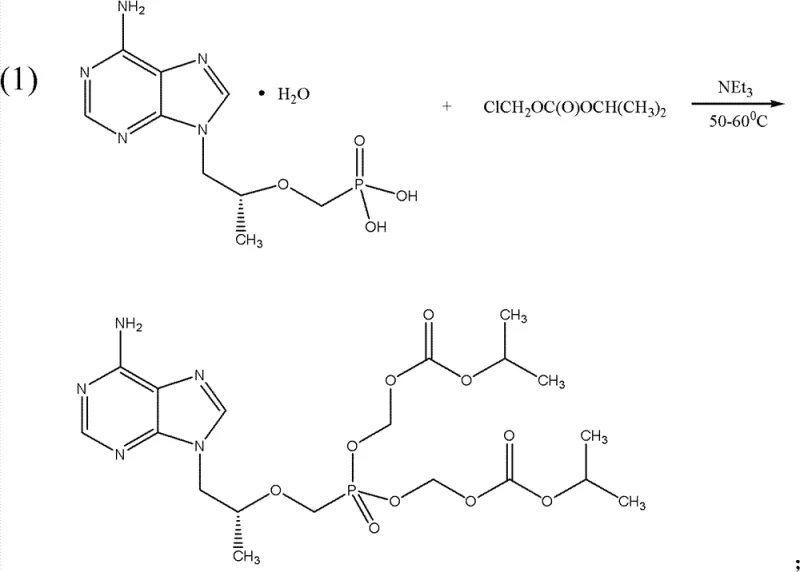
The methodology disclosed in CN103360425A represents a paradigm shift by eliminating the need for pre-reaction azeotropic dehydration entirely. Instead, the process employs solid dehydration agents such as anhydrous calcium chloride, anhydrous sodium sulfate, or anhydrous magnesium sulfate directly within the reaction matrix. This allows the condensation between Tenofovir (or its monohydrate) and chloromethyl isopropyl carbonate to proceed efficiently at moderate temperatures ranging from 50°C to 80°C. The substitution of problematic solvents with more manageable options like DMF or acetonitrile, coupled with the use of ethyl acetate for extraction, streamlines the workup procedure significantly. As illustrated in the reaction scheme below, this direct approach minimizes side reactions and simplifies the isolation of the intermediate tenofovir disoproxil, thereby enhancing the overall robustness of the synthetic route.
Mechanistic Insights into Condensation and Salt Formation
The core of this synthetic advancement lies in the precise control of the nucleophilic substitution reaction between the phosphonic acid group of tenofovir and the chloromethyl carbonate electrophile. In the presence of a tertiary amine base, such as triethylamine, the phosphonic acid is activated to facilitate the attack on the chloromethyl carbon, displacing the chloride ion. The inclusion of solid dehydration agents plays a dual mechanistic role: firstly, they sequester the water of crystallization from the tenofovir monohydrate reactant, preventing the hydrolysis of the sensitive chloromethyl isopropyl carbonate reagent; secondly, they help drive the equilibrium forward by removing water generated during the reaction, thus pushing the conversion towards the desired diester product. This mechanistic optimization ensures that the reaction proceeds with high selectivity, minimizing the formation of mono-ester impurities or hydrolyzed byproducts that are common in moisture-rich environments.
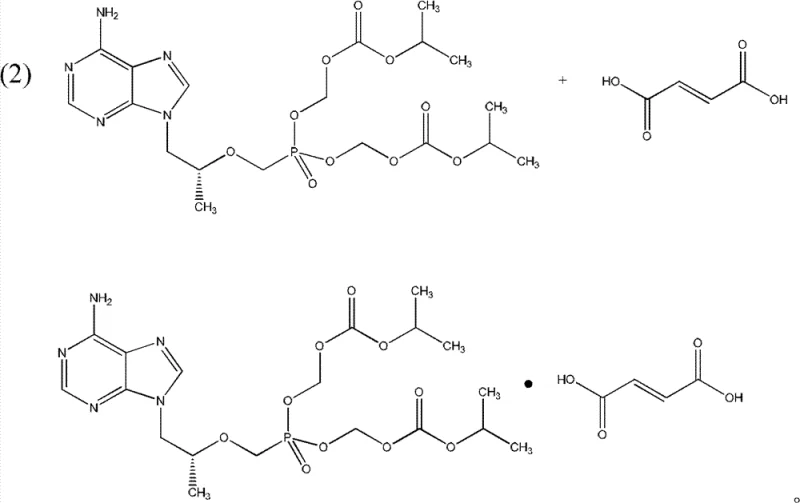
Following the successful synthesis of the tenofovir disoproxil intermediate, the process transitions to the critical salt formation step to generate the stable fumarate salt. The patent specifies a controlled dropping mode where the tenofovir disoproxil is added gradually to an alcoholic solution of fumaric acid maintained at 45°C to 55°C. This specific thermal window and addition rate are crucial for ensuring complete salification without inducing thermal degradation of the ester linkages. The choice of alcohol, preferably isopropanol or ethanol, facilitates the dissolution of fumaric acid while allowing the final fumarate salt to precipitate efficiently upon cooling. This controlled crystallization not only improves the physical properties of the powder, such as flowability and particle size distribution, but also ensures that the stoichiometric ratio of drug to acid is maintained within tight specifications, which is vital for the bioavailability and stability of the final dosage form.
How to Synthesize Tenofovir Disoproxil Efficiently
Implementing this optimized synthesis route requires careful attention to reagent quality and process parameters to maximize the benefits of the novel dehydration strategy. The protocol begins with the suspension of tenofovir monohydrate and a selected solid dehydration agent in a polar aprotic solvent, followed by the addition of the base and the alkylating agent. Maintaining the reaction temperature within the specified 50-80°C range is critical to balance reaction kinetics with the stability of the reagents. Upon completion, the workup involves a straightforward liquid-liquid extraction using ethyl acetate and water, which effectively partitions the organic product from inorganic salts and polar impurities. The detailed standardized operating procedures for scaling this reaction from laboratory to pilot plant are outlined in the technical guide below.
- Condense tenofovir monohydrate with chloromethyl isopropyl carbonate using a solid dehydration agent and triethylamine in DMF at 50-80°C.
- Extract the resulting tenofovir disoproxil using ethyl acetate and wash with water to remove inorganic salts and residual base.
- React the purified tenofovir disoproxil with fumaric acid in an alcoholic solution (e.g., isopropanol) at 45-55°C to form the final fumarate salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis method offers substantial strategic advantages beyond mere chemical yield improvements. By fundamentally altering the process architecture to remove energy-intensive distillation steps and hazardous solvents, the technology enables significant cost reduction in API manufacturing without compromising on quality standards. The elimination of azeotropic agents like hexane and toluene not only lowers raw material costs but also drastically reduces the expense associated with solvent recovery and waste disposal, contributing to a leaner and more sustainable production model. Furthermore, the simplified workflow reduces the total batch cycle time, allowing manufacturing facilities to increase throughput and respond more agilely to market demand fluctuations, thereby enhancing overall supply chain reliability.
- Cost Reduction in Manufacturing: The replacement of expensive and difficult-to-remove solvents like N-Methyl pyrrolidone (NMP) with more economical and volatile solvents like DMF and ethyl acetate results in substantial operational savings. The ability to use standard ethyl acetate for extraction instead of specialized low-boiling esters further drives down material costs, while the removal of the 24-hour azeotropic dehydration step significantly cuts energy consumption and labor hours, leading to a more competitive cost structure for the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route against moisture ingress eliminates a major source of batch failure and variability seen in conventional methods. By utilizing readily available solid dehydration agents and common organic solvents, the process reduces dependency on specialized reagents that may face supply constraints. This simplification of the bill of materials ensures a more stable and continuous supply of high-purity pharmaceutical intermediates, mitigating the risk of production delays caused by raw material shortages or complex equipment maintenance.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this method offers a cleaner profile by avoiding the use of large volumes of carcinogenic or neurotoxic solvents like hexane and benzene derivatives. The improved filterability of the product, due to the absence of viscous high-boiling solvents, facilitates easier scale-up in large reactors and reduces the burden on wastewater treatment facilities. This alignment with green chemistry principles not only ensures compliance with increasingly stringent environmental regulations but also enhances the corporate sustainability profile of the manufacturing partner.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis technology. These insights are derived directly from the experimental data and process descriptions found in the patent literature, providing a clear understanding of how this method compares to legacy processes. Understanding these nuances is critical for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the long-term value proposition of this manufacturing route.
Q: How does this method improve upon traditional azeotropic dehydration?
A: Traditional methods require lengthy azeotropic distillation with solvents like hexane or toluene to remove water, which is energy-intensive and risks moisture re-entry. This patent utilizes solid dehydration agents (e.g., anhydrous sodium sulfate) directly in the reaction mixture, significantly simplifying the operation and reducing processing time.
Q: What are the advantages regarding solvent removal and product purity?
A: Conventional processes often use high-boiling, viscous solvents like N-Methyl pyrrolidone (NMP), which are difficult to remove and lead to thick slurries that hinder filtration. This novel approach employs DMF or acetonitrile for reaction and ethyl acetate for extraction, ensuring easier solvent recovery, lower residual solvent levels, and superior filterability of the final product.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process is designed for scalability. By eliminating complex azeotropic setups and replacing expensive extraction agents with common solvents like ethyl acetate, the method reduces operational complexity and environmental impact, making it highly viable for commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tenofovir Disoproxil Fumarate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition to advanced synthesis methods like the one described in CN103360425A requires a partner with deep technical expertise and proven manufacturing capabilities. As a leading CDMO, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this novel dehydration and condensation strategy are fully realized in a GMP-compliant environment. Our state-of-the-art facilities are equipped to handle the specific solvent systems and temperature controls required for this process, and our rigorous QC labs enforce stringent purity specifications to guarantee that every batch of Tenofovir Disoproxil Fumarate meets the highest global pharmacopeial standards.
We invite potential partners to engage with our technical team to explore how this optimized synthesis route can be integrated into your supply chain to drive efficiency and quality. By requesting a Customized Cost-Saving Analysis, you can gain a detailed projection of the economic benefits specific to your volume requirements. We encourage you to contact our technical procurement team today to obtain specific COA data from our pilot batches and to discuss comprehensive route feasibility assessments tailored to your project timelines.