Scalable Synthesis of 6-Fluoro-3-Hydroxy-2-Pyrazinamide for Antiviral Drug Manufacturing
Introduction to Advanced Antiviral Intermediate Synthesis
The global demand for effective antiviral therapeutics has necessitated the development of robust, high-yield synthetic routes for key pharmaceutical intermediates. Among these, 6-fluoro-3-hydroxy-2-pyrazinamide, also known industrially as QD-Z0212, stands out as a critical building block for next-generation viral infection treatments. As detailed in the seminal patent CN102603658B, a novel preparation method has been established that overcomes the significant bottlenecks of previous generations of synthesis. This technology leverages a strategic four-step sequence involving amino protection, halogen displacement, deprotection, and azidation to deliver the target molecule with exceptional efficiency. By utilizing 6-bromo-3-amino-2-pyrazinamide as the starting material, the process ensures a streamlined workflow that is both economically viable and chemically elegant.
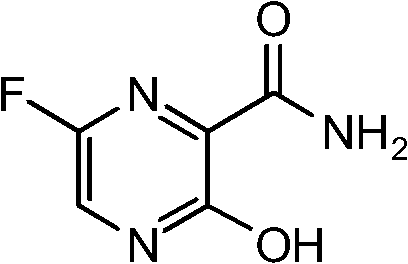
The structural integrity and purity of QD-Z0212 are paramount for its downstream application in drug formulation. The molecular architecture features a pyrazine core substituted with a fluorine atom at the 6-position, a hydroxyl group at the 3-position, and a carboxamide group at the 2-position. Achieving this specific substitution pattern without generating regio-isomers or halogenated impurities requires precise control over reaction conditions. The patented methodology addresses these challenges by introducing a temporary protecting group that shields the reactive amino functionality during the critical fluorination stage. This approach not only enhances the selectivity of the reaction but also simplifies the workup procedures, making it an ideal candidate for integration into modern continuous flow or batch manufacturing processes utilized by leading fine chemical producers.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of fluorinated pyrazine derivatives has been plagued by inefficient multi-step sequences that suffer from poor atom economy and hazardous reaction conditions. Traditional routes often commence with nitro-substituted precursors that require rigorous reduction steps followed by aggressive chlorination using phosphorus oxychloride (POCl3) under reflux. As illustrated in the comparative data associated with older methodologies, these pathways frequently involve the conversion of hydroxyl groups to chlorides, followed by a difficult nucleophilic substitution to reintroduce fluorine. Such transformations are notoriously capricious, often requiring anhydrous conditions and expensive catalysts to proceed with even moderate success. Furthermore, the final steps in these legacy processes, particularly the hydrolysis of nitrile groups or the displacement of halogens to form hydroxyl functionalities, have been documented to yield dismal results, with some isolated steps reporting yields as low as 26%.
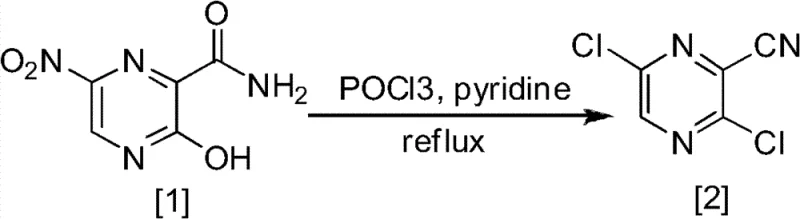
The cumulative effect of these low-yielding steps results in a catastrophic loss of material throughput, driving up the cost of goods sold (COGS) and creating significant waste disposal challenges. The reliance on harsh reagents like POCl3 also introduces severe safety risks regarding corrosion and exothermic runaway potential, complicating the regulatory approval for large-scale manufacturing. Additionally, the purification of intermediates in these conventional routes often necessitates column chromatography, a technique that is fundamentally incompatible with industrial-scale production due to solvent consumption and time inefficiencies. The presence of multiple halogenated byproducts further complicates the impurity profile, requiring extensive recrystallization efforts that erode the already thin profit margins associated with commodity intermediate production.
The Novel Approach
In stark contrast, the innovative process disclosed in patent CN102603658B revolutionizes the production landscape by adopting a protection-deprotection strategy that circumvents the pitfalls of direct halogenation. By initiating the synthesis with 6-bromo-3-amino-2-pyrazinamide, the route bypasses the need for dangerous nitro-reductions and aggressive chlorination entirely. The cornerstone of this novelty lies in the initial protection of the 3-amino group with a tert-butoxycarbonyl (Boc) moiety. This simple yet profound modification renders the molecule robust enough to withstand the subsequent nucleophilic aromatic substitution conditions required for fluorine installation. The result is a dramatic improvement in operational simplicity, where reactions can be conducted in standard polar aprotic solvents like DMF or DMSO at moderate temperatures ranging from 25°C to 60°C. This shift from extreme thermal and chemical stress to mild, controlled conditions represents a paradigm shift in process chemistry, enabling manufacturers to achieve consistent quality with significantly reduced operational expenditure.
Mechanistic Insights into Boc-Protection and Fluorination Strategy
The mechanistic elegance of this synthesis lies in the careful orchestration of electronic effects within the pyrazine ring system. In the first critical stage, the reaction of 6-bromo-3-amino-2-pyrazinamide with di-tert-butyl dicarbonate (Boc2O) in the presence of a base such as sodium hydride (NaH) or triethylamine creates a steric and electronic shield around the nitrogen atom. Without this protection, the free amino group would act as a competing nucleophile or undergo oxidation during the subsequent fluorination step, leading to complex polymeric tars and intractable mixtures. The formation of the N-Boc bond effectively lowers the electron density of the ring slightly while preventing side reactions, thereby priming the 6-position for selective attack. This step is typically executed in solvents like 1,4-dioxane or DMF, where the solubility of both the organic substrate and the inorganic base is optimized to ensure homogeneous reaction kinetics and complete conversion within a few hours.
Following protection, the installation of the fluorine atom proceeds via a classic nucleophilic aromatic substitution (SnAr) mechanism, facilitated by the electron-withdrawing nature of the pyrazine nitrogen atoms. The use of potassium fluoride (KF) as the fluorine source, often augmented by a phase transfer catalyst like tetrabutylammonium bromide, allows for the efficient displacement of the bromine leaving group. The bromine atom at the 6-position is activated by the adjacent ring nitrogens, making it susceptible to attack by the fluoride ion. The presence of the Boc group ensures that the reaction environment remains compatible with the basic fluoride source, preventing decomposition of the sensitive amide functionality. Experimental data from the patent indicates that this step can achieve yields upwards of 86%, a testament to the high selectivity of the protected intermediate. The subsequent acidic deprotection using hydrochloric acid cleanly removes the Boc group, regenerating the free amine without affecting the newly installed carbon-fluorine bond, which is kinetically stable under these acidic conditions.
How to Synthesize 6-Fluoro-3-Hydroxy-2-Pyrazinamide Efficiently
Implementing this synthesis in a GMP-compliant facility requires strict adherence to the optimized parameters outlined in the patent embodiments. The process is designed to be telescoped where possible, minimizing the isolation of intermediates to reduce handling losses and exposure to moisture. The initial protection step sets the tone for the entire sequence, requiring precise stoichiometric control of the base to prevent over-deprotonation which could lead to ring opening. Following the fluorination, the workup involves a straightforward aqueous extraction that leverages the differential solubility of the organic product versus inorganic salts. The final transformation, involving diazotization with sodium nitrite in sulfuric acid, must be conducted at low temperatures (-20°C to 10°C) to stabilize the diazonium intermediate before its hydrolysis to the phenol (hydroxyl) equivalent. For a detailed breakdown of the specific molar ratios, temperature gradients, and quenching protocols, please refer to the standardized operating procedure below.
- Protect the amino group of 6-bromo-3-amino-2-pyrazinamide using Boc2O and a base like NaH in DMF to form the t-butyl carbamate derivative.
- Perform nucleophilic aromatic substitution using Potassium Fluoride (KF) and a phase transfer catalyst in DMSO to replace the bromine atom with fluorine.
- Remove the Boc protecting group using concentrated hydrochloric acid in methanol to regenerate the free amino group at the 3-position.
- Convert the 3-amino group to a 3-hydroxyl group via diazotization with Sodium Nitrite in sulfuric acid followed by hydrolysis.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers tangible strategic advantages that extend far beyond simple chemical yield. The primary value proposition lies in the drastic simplification of the raw material supply chain. By utilizing 6-bromo-3-amino-2-pyrazinamide as the starting point, manufacturers can source materials from a broader range of suppliers who specialize in brominated heterocycles, rather than relying on scarce or geopolitically sensitive nitro-precursors. This diversification of the supply base inherently reduces the risk of production stoppages due to raw material shortages. Furthermore, the elimination of column chromatography purification steps means that the process is immediately scalable from pilot plant to full commercial production without the need for capital-intensive equipment upgrades. The ability to purify intermediates through simple crystallization or extraction significantly lowers the barrier to entry for contract manufacturing organizations (CMOs) looking to bid on production contracts.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the substantial increase in overall yield and the reduction in solvent and reagent consumption. By avoiding the low-yielding hydrolysis and chlorination steps characteristic of older methods, the mass balance of the reaction is preserved, meaning less starting material is wasted per kilogram of final product. The use of inexpensive inorganic salts like potassium fluoride and sodium bicarbonate, instead of exotic metal catalysts or anhydrous reagents, further drives down the variable cost of production. Additionally, the mild reaction temperatures reduce energy consumption for heating and cooling, contributing to a lower carbon footprint and reduced utility costs. These cumulative savings allow for a more competitive pricing structure in the global market for antiviral intermediates, providing a distinct margin advantage for manufacturers who license or adopt this technology.
- Enhanced Supply Chain Reliability: Reliability in the pharmaceutical supply chain is contingent upon the robustness of the manufacturing process. The novel route described in CN102603658B demonstrates exceptional tolerance to minor variations in reaction conditions, a critical factor for maintaining consistency across different batches and production sites. The intermediates generated, particularly the Boc-protected species, are stable and can be stored or transported if necessary, adding flexibility to the production schedule. This stability mitigates the risk of batch failures due to transient equipment issues or slight deviations in reagent quality. Consequently, suppliers can offer shorter lead times and more reliable delivery schedules to their downstream API clients, fostering stronger long-term partnerships and reducing the need for safety stock inventory which ties up working capital.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this process represents a significant advancement towards green chemistry principles. The avoidance of phosphorus oxychloride (POCl3) eliminates the generation of large volumes of acidic phosphorous waste, which is costly and difficult to treat. The solvent systems employed, such as DMSO and alcohols, are more amenable to recovery and recycling compared to the chlorinated solvents often required in traditional routes. The high selectivity of the reaction minimizes the formation of toxic halogenated byproducts, simplifying the wastewater treatment process. These factors not only ensure compliance with increasingly stringent environmental regulations but also enhance the corporate social responsibility (CSR) profile of the manufacturing entity, a key consideration for multinational pharmaceutical partners when selecting vendors.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of QD-Z0212. These insights are derived directly from the experimental data and claims presented in the patent literature, providing a transparent view of the technology's capabilities. Understanding these nuances is essential for R&D teams evaluating process transfer and procurement officers assessing vendor qualifications. The answers reflect the consensus on best practices for handling fluorinated pyrazine derivatives and highlight the specific advantages of the Boc-protection strategy.
Q: What are the critical yield improvements in the new synthesis route compared to conventional methods?
A: The novel four-step process described in patent CN102603658B significantly improves overall yield by avoiding harsh chlorination and low-efficiency hydrolysis steps found in older routes. Individual step yields consistently exceed 75%, with the fluorination step reaching up to 86%, ensuring a robust supply of the final API intermediate.
Q: How does the Boc-protection strategy enhance product purity?
A: By protecting the 3-amino group prior to fluorination, the process prevents unwanted side reactions and polymerization that typically occur under basic fluorination conditions. This strategic protection ensures that the nucleophilic substitution occurs selectively at the 6-position, drastically reducing impurity profiles and simplifying downstream purification.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the method utilizes readily available starting materials like 6-bromo-3-amino-2-pyrazinamide and common reagents such as KF and Boc2O. The reaction conditions are mild (25-60°C) and avoid the use of extremely hazardous reagents in large excess, making it highly adaptable for commercial scale-up from kilogram to multi-ton quantities.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6-Fluoro-3-Hydroxy-2-Pyrazinamide Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of antiviral drugs depends on a secure and high-quality supply of critical intermediates like 6-fluoro-3-hydroxy-2-pyrazinamide. Our technical team has extensively analyzed the pathway described in CN102603658B and integrated its core principles into our own manufacturing platforms. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the fluctuating demands of the global pharmaceutical market. Our facilities are equipped with state-of-the-art reactors capable of handling the specific temperature and pressure requirements of fluorination and diazotization reactions safely. We maintain stringent purity specifications and operate rigorous QC labs to ensure that every batch of QD-Z0212 meets the exacting standards required for API synthesis, minimizing the risk of downstream processing issues for our clients.
We invite potential partners to engage with our technical procurement team to discuss how our optimized manufacturing capabilities can support your drug development pipeline. By leveraging our expertise in process optimization, we can offer a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact us to request specific COA data and route feasibility assessments, allowing you to make informed decisions based on hard data rather than estimates. Together, we can accelerate the delivery of life-saving antiviral therapies to patients worldwide by establishing a resilient and efficient supply chain for this vital chemical building block.