Advanced Six-Step Synthesis of 6-Fluoro-3-Hydroxy-2-Pyrazinamide for Commercial Scale-Up
Advanced Six-Step Synthesis of 6-Fluoro-3-Hydroxy-2-Pyrazinamide for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust and scalable pathways for synthesizing complex heterocyclic intermediates, particularly those serving as key building blocks for antiviral agents. Patent CN102775358A discloses a highly efficient preparation method for 6-fluoro-3-hydroxy-2-pyrazinamide, also known as QD-Z0212, which is critical for treating viral infections including influenza. This technical disclosure represents a significant departure from legacy synthetic strategies by streamlining the production workflow into a concise six-step sequence that begins with the commercially accessible methyl 3-amino-2-pyrazinecarboxylate. By leveraging classical organic transformations such as diazotization, esterification, and catalytic hydrogenation, this methodology addresses the longstanding challenges of yield optimization and operational simplicity. For R&D directors and process chemists, understanding the nuances of this route provides a blueprint for achieving high-purity outputs without the burden of exotic reagents or extreme reaction parameters.
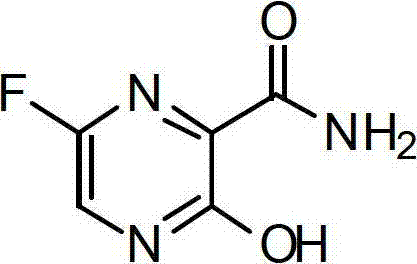
The strategic value of this patent lies not just in the final molecule but in the deliberate avoidance of kinetic traps that plague earlier syntheses. Traditional approaches often necessitated rigorous anhydrous environments and multiple protection-deprotection cycles that inherently degrade overall throughput. In contrast, the disclosed method operates under温和 (mild) conditions, utilizing aqueous workups and standard organic solvents that facilitate easier isolation of intermediates. This shift towards operational robustness is essential for supply chain heads who prioritize consistency and batch-to-batch reproducibility. Furthermore, the elimination of chromatographic purification steps in favor of crystallization and extraction drastically reduces solvent consumption and processing time, aligning perfectly with modern green chemistry initiatives and cost-reduction mandates in fine chemical manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as those described in WO0010569, typically rely on convoluted synthetic pathways that introduce unnecessary complexity and risk into the manufacturing process. A common historical strategy involved starting from 6-bromo-3-amino-2-pyrazine carboxylate methyl ester, which required a series of delicate transformations including diazotization followed by methoxy group introduction and subsequent Buchwald-Hartwig coupling to install the nitrogen functionality. These multi-step sequences are fraught with inefficiency; for instance, the protection and deprotection steps associated with introducing and removing methoxy groups during diazotization have been reported to yield as poorly as 35% and 15% respectively. Such low yields in critical intermediate stages compound exponentially, resulting in a dismal overall process mass intensity and inflated raw material costs. Additionally, these older routes often demand strictly anhydrous conditions and specialized reagents, creating significant bottlenecks for scale-up and increasing the hazard profile of the operation.
The Novel Approach
The innovative route presented in the patent data fundamentally reimagines the synthetic logic by prioritizing direct functionalization over protective group chemistry. By selecting methyl 3-amino-2-pyrazinecarboxylate as the starting scaffold, the process bypasses the need for halogen-metal exchanges or complex coupling reactions entirely. The pathway proceeds through a logical progression of hydroxylation, esterification, amidation, nitration, reduction, and finally fluorination, each step designed to maximize atom economy and minimize waste. As illustrated in the reaction scheme below, the transformation maintains the integrity of the pyrazine ring while systematically installing the required substituents with high regioselectivity. This streamlined approach not only shortens the operational path but also ensures that reaction conditions remain gentle and easily controllable, making it exceptionally well-suited for adaptation to industrial production equipment where stability and safety are paramount concerns.
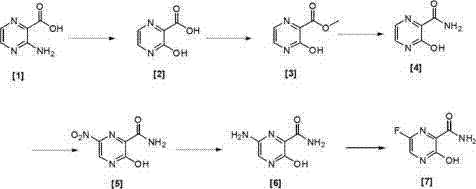
Mechanistic Insights into Diazotization and Fluorination Strategies
The cornerstone of this synthesis is the precise control of nitrogen-based functional group interconversions, specifically the initial hydroxylation and the terminal fluorination. The first step involves the diazotization of the primary amine on the pyrazine ring using sodium nitrite in a mineral acid solvent at temperatures below 0°C. This generates a reactive diazonium salt intermediate which, upon heating or spontaneous decomposition in the aqueous acidic medium, undergoes hydrolysis to yield the corresponding phenol-like hydroxyl group on the heterocycle. This transformation is critical because it establishes the 3-hydroxy motif early in the sequence, directing subsequent electrophilic substitutions and stabilizing the ring system against unwanted side reactions. The efficiency of this step, with reported yields reaching up to 90-96% in optimized embodiments, underscores the robustness of the diazotization protocol when applied to electron-deficient pyrazine systems.
The final fluorination step represents a sophisticated application of the Balz-Schiemann reaction variant, utilizing pyridine hydrofluoride (Py·HF) as both the solvent and the fluoride source. In this mechanism, the 6-amino precursor is subjected to diazotization again using sodium nitrite at low temperatures (0 to -20°C) to form the diazonium tetrafluoroborate-like species in situ. The presence of the fluoride ion from the pyridine complex facilitates the nucleophilic displacement of the nitrogen gas leaving group, effectively installing the fluorine atom at the 6-position. This method avoids the use of hazardous gaseous fluorine or expensive electrophilic fluorinating reagents, offering a safer and more cost-effective alternative for introducing fluorine into sensitive heterocyclic frameworks. The ability to perform this reaction in a homogeneous solution phase further enhances heat transfer and mixing, ensuring uniform conversion and minimizing the formation of defluorinated byproducts.
How to Synthesize 6-Fluoro-3-Hydroxy-2-Pyrazinamide Efficiently
Executing this synthesis requires careful attention to temperature control and stoichiometry, particularly during the exothermic diazotization and nitration phases. The process begins with the conversion of the starting amine to the hydroxy-acid, followed by esterification in methanol with sulfuric acid catalysis to protect the carboxylic acid as a methyl ester. Subsequent amidation with ammonia converts the ester to the primary amide, setting the stage for the introduction of the nitro group via mixed acid nitration. The nitro group is then selectively reduced to an amine using palladium on carbon catalysis under hydrogen pressure, preparing the molecule for the final fluorination event. While the general workflow is straightforward, the specific parameters regarding solvent ratios, acid concentrations, and cooling rates are critical for maximizing yield and purity. For a detailed breakdown of the standardized operating procedures and specific reagent quantities, please refer to the technical guide below.
- Hydroxylation of methyl 3-amino-2-pyrazinecarboxylate using sodium nitrite in mineral acid at low temperature to form 3-hydroxy-2-pyrazinecarboxylic acid.
- Esterification of the acid intermediate in methanol with sulfuric acid to yield the methyl ester derivative.
- Amidation reaction with ammonia in an organic solvent to convert the ester to 3-hydroxy-2-pyrazinecarboxamide.
- Nitration using concentrated nitric acid and sulfuric acid at sub-zero temperatures to introduce the nitro group at the 6-position.
- Catalytic hydrogenation using Pd/C in acidic methanol to reduce the nitro group to an amino group.
- Final fluorination using pyridine hydrofluoride and sodium nitrite to replace the amino group with fluorine, yielding the target compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the adoption of this synthetic route offers substantial advantages by decoupling production from scarce or expensive specialty reagents. The reliance on commodity chemicals such as sodium nitrite, sulfuric acid, methanol, and pyridine hydrofluoride ensures a stable and predictable supply chain, mitigating the risks associated with sourcing complex organometallic catalysts or custom-synthesized building blocks. This accessibility translates directly into cost stability, as the raw material basket for this process is composed of high-volume industrial chemicals that are less susceptible to market volatility. Furthermore, the simplification of the purification workflow—replacing column chromatography with crystallization and liquid-liquid extraction—significantly lowers the operational expenditure related to silica gel, eluents, and waste disposal, thereby enhancing the overall economic viability of the manufacturing campaign.
- Cost Reduction in Manufacturing: The elimination of low-yielding protection and deprotection steps inherently boosts the overall process yield, meaning less starting material is required to produce the same amount of final product. By avoiding the use of precious metal catalysts for coupling reactions and replacing them with standard heterogeneous hydrogenation catalysts like Pd/C, the process removes the need for expensive metal scavenging and recovery units. This reduction in catalyst cost, combined with the decreased solvent usage from simplified purification, results in a significantly lower cost of goods sold (COGS) for the final API intermediate.
- Enhanced Supply Chain Reliability: The use of readily available starting materials like methyl 3-amino-2-pyrazinecarboxylate ensures that production schedules are not held hostage by long lead times for custom precursors. The robustness of the reaction conditions, which tolerate mild variations in temperature and moisture better than anhydrous coupling protocols, reduces the likelihood of batch failures and reprocessing. This reliability allows supply chain managers to maintain leaner inventory levels while still meeting delivery commitments, as the risk of unexpected production stoppages due to reagent unavailability or sensitivity is drastically minimized.
- Scalability and Environmental Compliance: The process is explicitly designed for industrial adaptation, utilizing reaction vessels and separation techniques that are standard in multipurpose chemical plants. The avoidance of chlorinated solvents in favor of alcohols and esters, along with the reduction of heavy metal waste, simplifies the environmental permitting process and lowers the cost of wastewater treatment. The ability to run reactions at near-ambient temperatures or with simple ice-bath cooling reduces the energy load on the facility's HVAC and refrigeration systems, contributing to a smaller carbon footprint and aligning with increasingly stringent global sustainability regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthetic pathway. These answers are derived directly from the experimental data and comparative analysis provided in the patent documentation, focusing on practical considerations for process development. Understanding these details is crucial for teams evaluating the feasibility of technology transfer or scaling operations from pilot plant to commercial manufacturing volumes.
Q: What are the primary limitations of prior art synthesis methods for this compound?
A: Conventional methods often rely on complex multi-step sequences involving protection and deprotection strategies, such as introducing and removing methoxy groups via Buchwald-Hartwig coupling. These routes frequently suffer from low yields in critical steps (reported as low as 15-35%) and require harsh anhydrous conditions or expensive reverse-phase chromatography for purification.
Q: How does the new six-step route improve industrial feasibility?
A: The novel approach utilizes readily available starting materials like methyl 3-amino-2-pyrazinecarboxylate and avoids complex protecting group manipulations. The reaction conditions are significantly milder, often operating at ambient or near-ambient temperatures, and the process eliminates the need for column chromatography, relying instead on standard crystallization and extraction techniques suitable for large-scale manufacturing.
Q: What specific reagents are used for the final fluorination step?
A: The final transformation involves a diazotization-fluorination sequence where the 6-amino intermediate is treated with sodium nitrite in the presence of pyridine hydrofluoride solution. This serves as both the solvent and the fluorinating agent, allowing for the direct replacement of the diazonium group with fluorine under controlled low-temperature conditions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6-Fluoro-3-Hydroxy-2-Pyrazinamide Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory discovery to commercial reality requires a partner with deep technical expertise and unwavering commitment to quality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising yields observed in patent literature can be reliably replicated on an industrial scale. We operate state-of-the-art rigorous QC labs equipped to handle complex heterocyclic analysis, guaranteeing that every batch of 6-fluoro-3-hydroxy-2-pyrazinamide meets stringent purity specifications required for downstream pharmaceutical applications. Our infrastructure is designed to support the specific needs of fluorinated intermediates, with dedicated containment and safety protocols that manage the unique hazards associated with fluorination chemistry.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits specific to your volume requirements. We encourage you to reach out for specific COA data and route feasibility assessments, allowing us to demonstrate how our manufacturing capabilities can drive efficiency and reliability in your antiviral drug development programs. Let us collaborate to bring this critical intermediate to market faster and more cost-effectively.