Optimizing Ibrutinib Manufacturing: A Breakthrough in High-Purity Synthesis and Scalability
Optimizing Ibrutinib Manufacturing: A Breakthrough in High-Purity Synthesis and Scalability
The pharmaceutical landscape for oncology treatments continues to evolve, with Ibrutinib standing as a cornerstone tyrosine kinase inhibitor for treating mantle cell lymphoma and chronic lymphocytic leukemia. However, the commercial viability of this critical active pharmaceutical ingredient (API) has historically been constrained by complex purification challenges and persistent impurity profiles. Patent CN112125913A introduces a transformative synthesis method that addresses these bottlenecks through a novel acylation strategy. This technical insight report analyzes the proprietary methodology which achieves high-purity Ibrutinib (>99%) by fundamentally altering the reaction sequence to suppress the formation of difficult-to-remove side products. By shifting from traditional direct acylation to a pre-activated intermediate approach, manufacturers can significantly streamline production workflows.
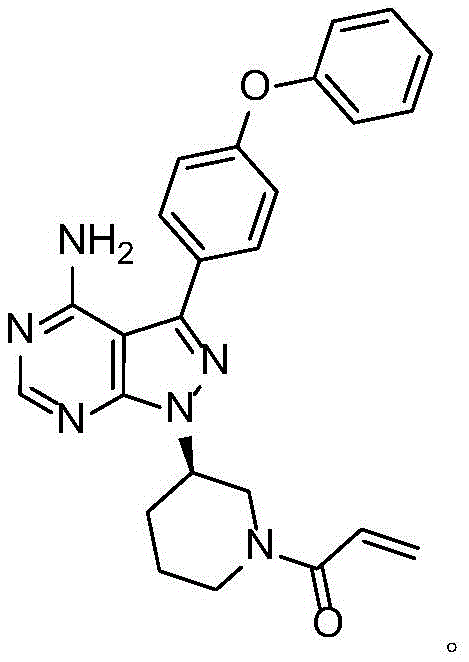
The structural integrity of the final molecule is paramount for biological efficacy, and the method described in CN112125913A ensures that the delicate pyrazolo-pyrimidine scaffold remains intact while the acrylamide tail is installed with precision. This innovation represents a significant leap forward for reliable API intermediate suppliers seeking to enhance their portfolio with robust, scalable chemistry. The following analysis dissects the mechanistic advantages, operational parameters, and commercial implications of this refined synthetic route.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for Ibrutinib often suffer from poor selectivity during the final acylation step, leading to the generation of structurally related impurities that are notoriously difficult to purge. Specifically, the formation of Impurity V, a chloropropionyl-derived byproduct, poses a severe challenge to process chemists and quality control teams. In standard protocols where the acylating agent is added directly to the reaction mixture containing the amine and base, localized exotherms and uncontrolled reaction kinetics frequently drive the formation of this stable impurity. Once formed, Impurity V exhibits physicochemical properties similar to the target API, necessitating multiple, yield-depleting recrystallization steps or expensive chromatographic purification to meet regulatory specifications. This not only inflates the cost of goods sold (COGS) but also extends the manufacturing lead time, creating bottlenecks in the supply chain for high-purity pharmaceutical intermediates.
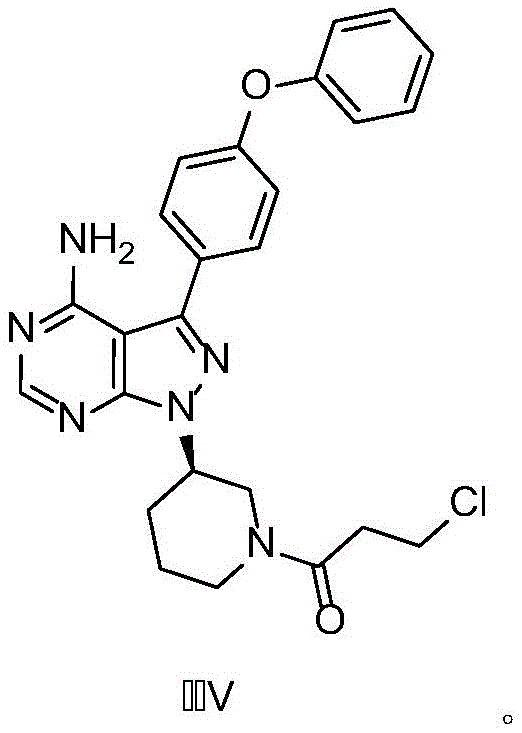
The Novel Approach
The methodology disclosed in CN112125913A circumvents these issues by decoupling the activation of the acylating agent from the nucleophilic attack. Instead of a one-pot mixture, the process involves the pre-reaction of an acyl halide derivative (Compound III) with a basic compound (Compound IV) to generate a stabilized acylation solution prior to the introduction of the core intermediate (Formula II). This "pre-mixing" strategy effectively moderates the reactivity of the acyl halide, preventing the harsh conditions that typically lead to the chloropropionyl side reactions responsible for Impurity V. By controlling the stoichiometry and thermal profile of this pre-activation step, the process ensures that when the core amine is introduced, the acylation proceeds with high fidelity. This approach eliminates the need for extensive downstream purification, thereby facilitating industrial large-scale production with consistently high purity profiles exceeding 99%.
Mechanistic Insights into Controlled Acylation Kinetics
The core innovation of this synthesis lies in the kinetic control of the acylation event. In the conventional pathway, the simultaneous presence of the amine, base, and acyl chloride creates a competitive environment where the base may inadvertently catalyze side reactions or where the acyl chloride undergoes hydrolysis or rearrangement before reacting with the amine. The novel method utilizes a specific molar ratio of acyl halide to base (preferably 1:1.2) to form a transient, activated complex. When this complex encounters the nucleophilic nitrogen of the piperidine ring in Formula II, the energy barrier for the desired substitution is lowered relative to competing pathways. The use of polar aprotic solvents such as N,N-dimethylacetamide (DMAc) or acetonitrile further stabilizes the transition state, enhancing the solubility of the intermediates and ensuring homogeneous reaction conditions. This precise orchestration of reagents minimizes the residence time of reactive species that could otherwise degrade into Impurity V.
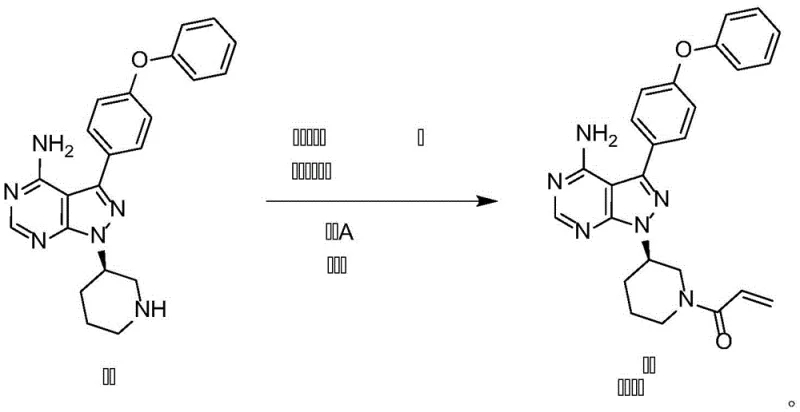
Furthermore, the temperature control parameters specified in the patent—ranging from -10°C to 30°C during the activation phase and -10°C to 70°C during the coupling phase—are critical for maintaining this kinetic selectivity. Low-temperature pre-mixing (-5°C is preferred) suppresses the thermal degradation of the acyl halide, while the subsequent controlled addition to the amine solution prevents localized hot spots that could trigger runaway side reactions. The result is a clean reaction profile where the primary outcome is the formation of the acrylamide bond, with negligible generation of the chlorinated impurity. This mechanistic understanding allows process engineers to design reactors with appropriate cooling capacities and dosing rates to replicate this high-selectivity environment on a multi-ton scale, ensuring batch-to-batch consistency essential for regulatory compliance.
How to Synthesize High-Purity Ibrutinib Efficiently
Implementing this synthesis route requires strict adherence to the sequential addition of reagents and temperature management protocols outlined in the patent data. The process begins with the preparation of the activated acylating species in a dedicated vessel, followed by its controlled transfer to the reactor containing the dissolved intermediate. This two-stage approach distinguishes the method from simpler one-pot procedures and is the key to achieving the reported purity levels of >99% and yields exceeding 88%. The final isolation step utilizes water as an anti-solvent, inducing crystallization directly from the reaction mixture, which simplifies the workup procedure significantly. For detailed operational parameters, including specific solvent volumes, dropwise addition rates, and stirring speeds required to replicate these results in a pilot or production plant, refer to the standardized protocol below.
- Pre-react acyl halide compound III with a basic compound IV in a suitable solvent at low temperatures (-10 to 30°C) to form an activated acylation solution.
- Disperse the core pyrazolo-pyrimidine intermediate (Formula II) in solvent and slowly add the activated acylation solution while maintaining strict temperature control.
- Quench the reaction mixture with purified water to induce crystallization, followed by filtration and drying to isolate Ibrutinib with >99% purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis method translates into tangible operational efficiencies and risk mitigation. The primary value driver is the drastic simplification of the purification train. By effectively inhibiting the generation of Impurity V at the source, the process eliminates the need for repetitive recrystallizations or resource-intensive chromatographic separations that are typical of legacy routes. This reduction in unit operations directly correlates to a significant decrease in processing time and solvent consumption, lowering the overall environmental footprint and utility costs associated with manufacturing. Furthermore, the use of widely available, commodity-grade reagents such as acryloyl chloride and common organic bases ensures that raw material sourcing remains stable and cost-effective, shielding the supply chain from volatility associated with exotic catalysts or specialized reagents.
- Cost Reduction in Manufacturing: The elimination of complex purification steps results in substantial cost savings by reducing solvent usage, energy consumption for distillation, and labor hours required for multiple isolation cycles. The high conversion rate and yield (>88%) mean that less starting material is wasted, optimizing the atom economy of the process. Additionally, the ability to use water for the final crystallization step reduces the reliance on expensive organic anti-solvents, further driving down the variable costs per kilogram of produced API. These factors combine to create a more economically viable production model that enhances margin potential without compromising on quality standards.
- Enhanced Supply Chain Reliability: The robustness of this chemical route ensures consistent output quality, which is critical for maintaining uninterrupted supply to downstream formulation partners. Because the process is less sensitive to minor fluctuations in reaction conditions due to the pre-activation step, the risk of batch failure is minimized. This reliability allows for more accurate production planning and inventory management, reducing the need for excessive safety stock. Moreover, the scalability of the method means that production capacity can be ramped up quickly to meet surges in market demand for Ibrutinib, providing a strategic advantage in a competitive therapeutic area.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, the process offers distinct advantages. The mild reaction temperatures reduce the thermal load on plant infrastructure, and the reduced solvent volume lowers the burden on waste treatment facilities. The substitution of hazardous purification solvents with water for crystallization aligns with green chemistry principles, facilitating easier regulatory approval and permitting. The simplicity of the workflow also reduces the potential for operator error during scale-up, ensuring that the transition from laboratory bench to commercial reactor is smooth and compliant with current Good Manufacturing Practices (cGMP).
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this high-purity synthesis method. These insights are derived directly from the experimental data and comparative examples provided in the patent literature, offering clarity on critical process parameters and quality outcomes. Understanding these nuances is essential for technical teams evaluating the feasibility of technology transfer or process validation.
Q: How does this synthesis method effectively control Impurity V?
A: By pre-mixing the acyl halide with the base before introducing the core intermediate, the reaction kinetics are optimized to favor the desired N-acylation while suppressing the formation of the chloropropionyl-derived side product known as Impurity V.
Q: What are the preferred solvents and bases for this process?
A: The patent identifies N,N-dimethylacetamide (DMAc) and acetonitrile as preferred solvents. For bases, organic amines like DIPEA or DBU (1,8-diazabicycloundecen-7-ene) provide superior results compared to inorganic carbonates.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the method utilizes mild reaction conditions (-10 to 30°C), common industrial solvents, and a simple water-induced crystallization step, making it highly amenable to commercial scale-up without complex purification requirements.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ibrutinib Supplier
At NINGBO INNO PHARMCHEM, we recognize that the complexity of modern oncology APIs demands a partner with deep technical expertise and unwavering commitment to quality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from lab-scale optimization to full-scale manufacturing is seamless. We leverage advanced analytical capabilities in our rigorous QC labs to verify stringent purity specifications, guaranteeing that every batch of Ibrutinib meets the highest international standards. Our facility is equipped to handle the precise temperature controls and specialized reagent handling required by the CN112125913A protocol, delivering a product that is ready for immediate formulation.
We invite global pharmaceutical partners to engage with our technical procurement team to discuss how this advanced synthesis route can optimize your supply chain. By requesting a Customized Cost-Saving Analysis, you can quantify the potential economic benefits of switching to this high-efficiency method. We encourage you to contact us today to obtain specific COA data and comprehensive route feasibility assessments tailored to your project timelines. Let us collaborate to bring high-quality, life-saving medications to patients faster and more efficiently.