Advanced Manufacturing of Delgocitinib Intermediate via Protection-Free Cyclization Strategy
The pharmaceutical landscape for atopic dermatitis treatment has been significantly advanced by the approval of Delgocitinib, a pan-JAK inhibitor. However, the commercial viability of such potent small molecules often hinges on the efficiency of their supply chain, specifically the synthesis of complex intermediates. Patent CN111560021A discloses a groundbreaking preparation method for the key Delgocitinib intermediate, (3S,4R)-3-methyl-beta-oxo-1,6-diazaspiro[3,4]octane-1-propionitrile. This technology represents a paradigm shift from traditional multi-step protection strategies to a more direct, green chemical synthesis approach. By utilizing common compounds as starting materials and sequentially executing reactions such as bromination, acylation, and cyclization, this method addresses the critical bottlenecks of yield loss and high operational costs associated with legacy routes. For R&D directors and procurement specialists, understanding this novel pathway is essential for securing a reliable pharmaceutical intermediate supplier capable of delivering high-purity materials at a competitive cost structure.
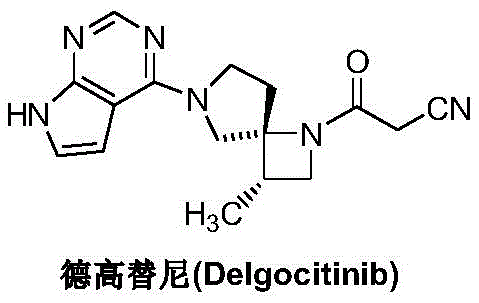
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of the diazaspiro core found in Delgocitinib has been plagued by inefficiencies inherent to protection group chemistry. International patents such as WO2011013785 and WO2018117152 describe routes that rely heavily on the mono- or double-protection of nitrogen atoms within the spiro-intermediate using benzyl or formyloxybenzyl groups. While these protecting groups serve to distinguish the reactivity of the two nitrogen atoms and minimize side reactions, they introduce substantial synthetic overhead. The requirement for subsequent deprotection steps not only adds at least two to three additional unit operations but also necessitates the use of harsh reagents and generates significant chemical waste. Furthermore, each additional step in a linear synthesis inevitably erodes the overall yield, making the cost reduction in API manufacturing difficult to achieve. The reliance on these cumbersome protection-deprotection sequences creates a fragile supply chain vulnerable to raw material fluctuations and extended lead times.
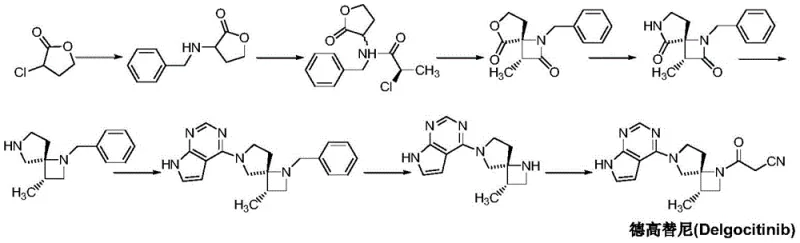
The Novel Approach
In stark contrast to the convoluted legacy pathways, the methodology outlined in CN111560021A offers a streamlined, protection-free strategy that radically simplifies the construction of the spiro-framework. This novel approach leverages the intrinsic reactivity differences of the functional groups to drive the cyclization without the need for temporary masking agents. By starting with R-2-chloro-1-propylamine and progressing through a carefully orchestrated sequence of acylation and base-mediated cyclization, the process achieves the target architecture with remarkable efficiency. The elimination of benzyl protection steps translates directly into a shorter critical path for production, enhancing the commercial scale-up of complex pharmaceutical intermediates. This route not only improves the economic feasibility of the drug substance but also aligns with modern green chemistry principles by reducing solvent consumption and waste generation, thereby offering a robust solution for high-purity API intermediate production.
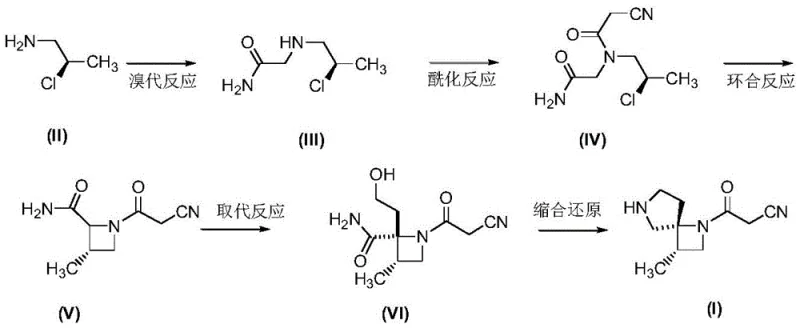
Mechanistic Insights into Cs2CO3-Mediated Cyclization and Condensation
The cornerstone of this innovative synthesis lies in the precise control of reactivity during the ring-closing and functionalization stages. The cyclization step, converting the acyclic N-(R-2-chloro-propyl)-N-(2-acetamide)-2-cyanoacetamide into the azetidine-containing intermediate, is facilitated by cesium carbonate in dimethyl sulfoxide. The choice of cesium carbonate is critical; its large cation radius enhances solubility in organic media and acts as a mild yet effective base to promote intramolecular nucleophilic substitution without compromising the sensitive nitrile or amide functionalities. This step establishes the crucial 1,6-diazaspiro[3,4]octane scaffold with high stereochemical fidelity, preserving the chirality introduced by the starting R-2-chloro-1-propylamine. Following cyclization, the introduction of the hydroxyethyl side chain via substitution with 2-bromoethanol and lithium hexamethyldisilazide (LiHMDS) demonstrates excellent regioselectivity. The use of LiHMDS, a strong non-nucleophilic base, ensures deprotonation occurs selectively at the desired nitrogen center, preventing oligomerization or unwanted side reactions that could compromise the purity profile of the intermediate.
Furthermore, the final transformation involves a sophisticated condensation-reduction sequence utilizing diethyl azodicarboxylate (DEAD), triphenylphosphine, and lithium aluminum hydride. This cascade effectively converts the hydroxyethyl precursor into the final propionitrile side chain while simultaneously closing the second ring of the spiro system. The mechanism likely proceeds through a Mitsunobu-type activation of the alcohol followed by intramolecular displacement and subsequent reduction of the resulting intermediate. This one-pot or telescoped strategy minimizes the isolation of unstable intermediates, thereby reducing material loss and exposure to environmental factors. From an impurity control perspective, the robustness of these reaction conditions ensures that by-products are minimized, simplifying downstream purification. For quality assurance teams, this mechanistic clarity provides confidence in the consistency of the batch-to-batch quality, a vital factor for regulatory compliance in the production of dermatological therapeutics.
How to Synthesize Delgocitinib Intermediate Efficiently
The implementation of this synthesis route requires strict adherence to the optimized reaction parameters defined in the patent to ensure maximum yield and safety. The process begins with the bromination of the chiral amine in n-hexane, followed by acylation in dichloromethane, setting the stage for the critical cyclization. Operators must maintain precise temperature controls, particularly during the low-temperature substitution step (-70 to -75°C), to prevent racemization or decomposition. The detailed standardized synthesis steps, including specific molar ratios and workup procedures, are outlined below to guide process engineers in replicating this high-efficiency protocol.
- Perform bromination of R-2-chloro-1-propylamine with 2-bromoacetamide using potassium carbonate in n-hexane to form the aminoacetamide precursor.
- Execute acylation with 2-cyanoacetyl chloride followed by cesium carbonate-mediated cyclization to construct the azetidine ring system.
- Complete the synthesis via lithium hexamethyldisilazide substitution and subsequent condensation-reduction using DEAD/PPh3 and LiAlH4.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route offers tangible strategic benefits that extend beyond mere technical elegance. The primary advantage lies in the drastic simplification of the manufacturing workflow, which directly correlates to reduced operational expenditures and enhanced supply reliability. By removing the protection and deprotection stages, the process eliminates the need for expensive protecting group reagents such as benzyl bromide or chloroformates, as well as the catalysts required for their removal. This reduction in material intensity significantly lowers the variable cost per kilogram of the intermediate. Moreover, the shortened process flow reduces the turnaround time for production batches, allowing for greater flexibility in responding to market demand fluctuations. The use of common, commodity-grade starting materials further insulates the supply chain from the volatility often associated with specialized fine chemical building blocks, ensuring a steady and predictable flow of materials for continuous manufacturing campaigns.
- Cost Reduction in Manufacturing: The elimination of protection-deprotection sequences removes entire unit operations from the production schedule, leading to substantial savings in labor, energy, and solvent usage. Without the need to install and remove bulky benzyl groups, the process avoids the costly hydrogenation or acidic hydrolysis steps typically required, which often involve expensive catalysts like palladium on carbon or large volumes of corrosive acids. This streamlining results in a leaner manufacturing process with a smaller physical footprint and lower utility consumption, driving down the overall cost of goods sold (COGS) for the API. Additionally, the higher cumulative yield achieved by skipping multiple steps means that less raw material is wasted, maximizing the output from every kilogram of input and improving the economic viability of the project.
- Enhanced Supply Chain Reliability: The reliance on readily available starting materials such as R-2-chloro-1-propylamine and 2-bromoacetamide ensures that the supply chain is not dependent on single-source suppliers for exotic reagents. These commodity chemicals are produced at scale by multiple global manufacturers, reducing the risk of supply disruptions due to geopolitical issues or production outages at a specific vendor. The robustness of the reaction conditions, which tolerate standard industrial solvents like toluene, n-hexane, and tetrahydrofuran, further simplifies logistics and inventory management. This accessibility allows procurement teams to negotiate better pricing and secure long-term contracts, guaranteeing the continuity of supply necessary for meeting clinical and commercial launch timelines without the fear of bottlenecking on critical intermediates.
- Scalability and Environmental Compliance: The green chemistry attributes of this route facilitate easier regulatory approval and environmental compliance, which are increasingly critical for sustainable manufacturing. By reducing the number of reaction steps and avoiding heavy metal catalysts in the main synthetic line, the process generates less hazardous waste, lowering the costs associated with waste treatment and disposal. The scalability of the cyclization and substitution steps has been demonstrated to be robust, with exotherms that can be managed effectively in large-scale reactors. This ease of scale-up from laboratory to commercial production (100 kgs to 100 MT) minimizes the technical risks usually encountered during technology transfer, ensuring that the transition from pilot plant to full-scale manufacturing is smooth and predictable, thereby safeguarding the project timeline.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of this Delgocitinib intermediate. These answers are derived directly from the experimental data and technical specifications provided in the patent literature, offering clarity on the feasibility and advantages of this specific synthetic pathway for industry stakeholders.
Q: How does this new route improve upon previous benzyl-protection methods?
A: The novel route eliminates the need for benzyl or formyloxybenzyl protection and deprotection steps, significantly reducing the total number of reaction stages and improving overall atom economy.
Q: What are the critical reaction conditions for the cyclization step?
A: The cyclization of the chloro-propyl intermediate requires cesium carbonate as a base in dimethyl sulfoxide (DMSO) at temperatures between 35-45°C to ensure high yield and stereochemical integrity.
Q: Is the starting material commercially available for scale-up?
A: Yes, the synthesis begins with R-2-chloro-1-propylamine and 2-bromoacetamide, which are common, economically viable industrial chemicals, facilitating easy procurement for large-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Delgocitinib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient intermediate synthesis in the race to bring life-changing dermatological treatments to market. Our team of expert chemists has thoroughly analyzed the protection-free route described in CN111560021A and validated its potential for industrial application. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop discovery to commercial reality is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of Delgocitinib intermediate we supply meets the highest international standards for safety and efficacy. We are committed to being a partner that not only supplies chemicals but also delivers technical excellence and supply chain security.
We invite pharmaceutical companies and contract research organizations to collaborate with us to leverage this advanced manufacturing technology. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this optimized route can improve your bottom line. We encourage you to contact us today to discuss your project needs,索取 specific COA data for our reference standards, and review our comprehensive route feasibility assessments. Let us help you secure a competitive edge in the JAK inhibitor market with our reliable, high-quality, and cost-effective intermediate solutions.