Advanced Synthesis of Delgocitinib Intermediate via Green Chemical Pathway for Commercial Scale-up
Introduction to the Novel Delgocitinib Intermediate Synthesis
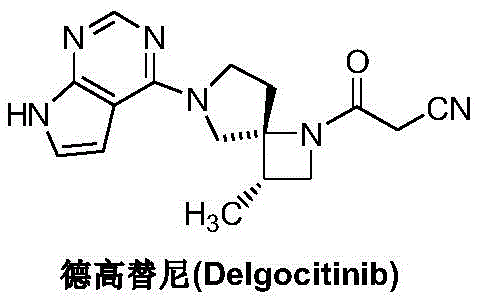
The pharmaceutical landscape for JAK kinase inhibitors has been significantly advanced by the development of Delgocitinib, a potent therapeutic agent for atopic dermatitis. As detailed in patent CN111732595A, a groundbreaking approach has been established for the preparation of its critical precursor, the (3S,4R)-3-methyl-beta-oxo-1,6-diazaspiro[3,4]octane-1-propionitrile intermediate. This innovation addresses long-standing challenges in the synthetic chemistry of spiro-cyclic compounds, offering a streamlined pathway that bypasses the inefficiencies of traditional multi-step protection strategies. By leveraging common, commercially available starting materials and a sequence of robust reactions including bromination, acylation, and cyclization, this method provides a sustainable and economically viable route for high-purity pharmaceutical intermediate production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
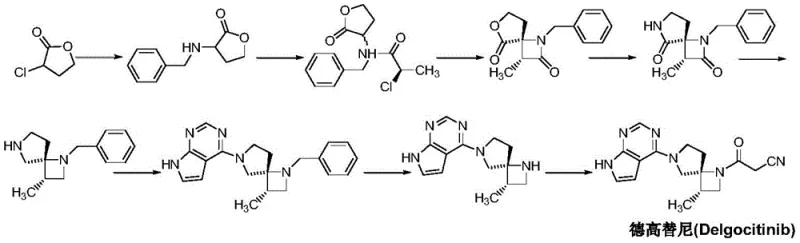
Historically, the synthesis of complex spiro-intermediates like those required for Delgocitinib has been plagued by inefficient protection-deprotection sequences. Prior art, such as international patents WO2011013785 and WO2018117152, discloses routes that rely heavily on mono- or double-protection of nitrogen atoms using benzyl or formyloxybenzyl groups. While these protecting groups serve to differentiate the reactivity of the two nitrogen centers and minimize side reactions, they introduce significant operational burdens. The necessity to install and subsequently remove these bulky groups inherently increases the number of synthetic steps, which invariably leads to a cumulative loss in overall yield. Furthermore, the reagents required for deprotection often involve harsh conditions or expensive catalysts, driving up the cost of goods sold (COGS) and generating substantial chemical waste that complicates environmental compliance.
The Novel Approach
In stark contrast, the methodology presented in CN111732595A revolutionizes this landscape by eliminating the need for nitrogen protection entirely. The new strategy utilizes a cleverly designed sequence where the reactivity of the amine and the nitrile groups is managed through precise stoichiometric control and specific reaction conditions rather than steric blocking. This approach not only truncates the synthetic timeline but also enhances the atom economy of the process. By avoiding the installation of benzyl groups, the process sidesteps the need for hydrogenolysis or acidic hydrolysis steps, thereby reducing the risk of racemization and preserving the critical (3S,4R) stereochemistry essential for biological activity. This represents a paradigm shift towards greener chemistry, aligning perfectly with the goals of modern cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into the Cascade Cyclization and Substitution Strategy
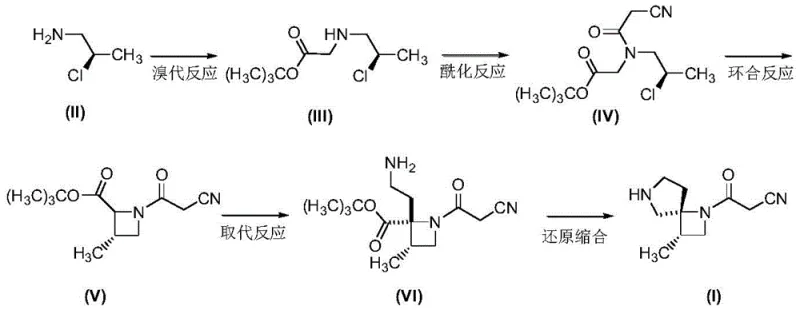
The core of this synthetic breakthrough lies in the sequential construction of the diazaspiro skeleton through a series of highly controlled transformations. The process initiates with a nucleophilic substitution where R-2-chloro-1-propylamine reacts with tert-butyl 2-bromoacetate under basic conditions to form an amino ester. This is followed by an acylation with 2-cyanoacetyl chloride, introducing the nitrile functionality required for the subsequent ring closure. The pivotal moment in the synthesis is the intramolecular cyclization mediated by cesium carbonate in dimethyl sulfoxide. The choice of cesium carbonate is critical; its large cation radius and moderate basicity facilitate the deprotonation of the active methylene group adjacent to the nitrile, promoting a clean SN2 attack on the chloro-substituted carbon to close the four-membered azetidine ring with high stereoselectivity.
Following the formation of the azetidine core, the synthesis proceeds through a substitution reaction with 2-bromoethylamine, utilizing hexamethyldisilazane lithium amide (LiHMDS) as a strong, non-nucleophilic base to ensure selective alkylation without disrupting the sensitive ester or nitrile moieties. The final transformation involves a reductive condensation cascade. Here, lithium aluminum hydride reduces the ester to an alcohol or aldehyde equivalent in situ, which then undergoes condensation with the pendant amine to close the second ring, forming the spiro-system. The use of triphenylphosphine and carbon tetrachloride in the final stage likely facilitates an Appel-type activation or similar dehydration mechanism to finalize the spiro-structure, ensuring the formation of the stable 1,6-diazaspiro[3,4]octane framework with the requisite beta-oxo-propionitrile side chain intact.
How to Synthesize Delgocitinib Intermediate Efficiently
The execution of this synthesis requires strict adherence to the optimized reaction parameters outlined in the patent to ensure maximum yield and purity. The process is divided into five distinct operational stages, each building upon the previous intermediate without the need for isolation of unstable species where possible. The initial bromination and acylation steps set the foundation for the carbon backbone, while the cyclization step establishes the core chirality. For R&D teams looking to implement this, the key lies in the precise control of temperature during the substitution phase (-70 to -75°C) to prevent over-alkylation, and the careful quenching of the reductive condensation to avoid decomposition of the spiro-ring. Detailed standardized operating procedures for each step, including specific workup protocols like aqueous washing and recrystallization, are essential for reproducibility.
- Perform bromination of R-2-chloro-1-propylamine with tert-butyl 2-bromoacetate using potassium carbonate in n-hexane at 60-70°C to form the amino ester intermediate.
- Conduct acylation with 2-cyanoacetyl chloride and triethylamine in dichloromethane, followed by cyclization using cesium carbonate in DMSO to form the azetidine ring.
- Execute substitution with 2-bromoethylamine using LiHMDS, followed by reductive condensation with lithium aluminum hydride and subsequent cyclization to yield the final spiro-intermediate.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route offers compelling strategic advantages that extend beyond simple technical feasibility. The elimination of protection and deprotection steps translates directly into a drastic simplification of the supply chain, as there is no longer a need to source specialized protecting group reagents or manage the logistics of their disposal. This streamlining reduces the number of unit operations required in the manufacturing plant, effectively increasing throughput capacity without the need for additional capital expenditure on equipment. Furthermore, the use of common solvents such as n-hexane, dichloromethane, and tetrahydrofuran ensures that raw material sourcing remains stable and resilient against market fluctuations, securing the continuity of supply for downstream API production.
- Cost Reduction in Manufacturing: The most significant economic benefit arises from the reduction in step count. By removing the benzyl protection and deprotection stages, the process saves on reagent costs, solvent consumption, and energy usage associated with heating and cooling cycles for extra steps. Additionally, the simplified workup procedures, which rely on standard filtration and washing rather than complex chromatographic purifications often needed for protected intermediates, significantly lower labor and processing costs. This lean manufacturing approach results in a substantially lower cost per kilogram of the final intermediate, enhancing the overall margin profile for the bulk drug.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals like R-2-chloro-1-propylamine and tert-butyl 2-bromoacetate mitigates the risk of supply bottlenecks. Unlike specialized chiral auxiliaries or exotic catalysts that may have single-source suppliers, these starting materials are widely produced by multiple chemical manufacturers globally. This diversification of the supply base ensures that production schedules are not held hostage by vendor shortages, providing a robust buffer against global logistical disruptions and guaranteeing consistent delivery timelines for pharmaceutical partners.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this route is markedly superior. The avoidance of heavy metal catalysts and the reduction in total solvent volume per unit of product decrease the facility's environmental footprint. The waste streams generated are simpler to treat, as they lack the complex organic residues associated with benzyl deprotection byproducts. This ease of waste management facilitates smoother regulatory approvals for commercial scale-up, allowing manufacturers to ramp up production from pilot batches to multi-ton annual capacities with greater speed and lower compliance risks.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, aiming to clarify the practical implications for industrial adoption. Understanding these nuances is crucial for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the long-term value proposition of this manufacturing method.
Q: How does this new synthesis route improve upon conventional methods for Delgocitinib intermediates?
A: Conventional routes typically require cumbersome benzyl or formyloxybenzyl protection and deprotection steps to distinguish nitrogen activities. This novel method eliminates those protection groups entirely, significantly reducing reaction steps, improving total yield, and lowering production costs while using common, economically viable starting materials.
Q: What are the key reaction conditions for the critical cyclization step?
A: The cyclization reaction, which forms the core azetidine structure, utilizes cesium carbonate as the base in dimethyl sulfoxide (DMSO) solvent. The reaction is optimally conducted at a temperature range of 35-45°C, ensuring high conversion rates and stereochemical integrity without the need for extreme conditions.
Q: Is this process suitable for large-scale industrial manufacturing?
A: Yes, the process is designed with industrial scalability in mind. It employs common solvents like n-hexane, dichloromethane, and THF, and avoids expensive transition metal catalysts or hazardous reagents that complicate waste treatment. The simplified workup procedures, such as filtration and standard aqueous washing, facilitate easier scale-up from pilot to commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Delgocitinib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthetic routes in the competitive landscape of JAK inhibitor production. Our team of expert chemists has thoroughly analyzed the methodology described in CN111732595A and is fully prepared to execute this advanced synthesis at scale. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless. Our facilities are equipped with stringent purity specifications and rigorous QC labs capable of monitoring every critical parameter, from stereochemical integrity to trace impurity profiling, guaranteeing a product that meets the highest global pharmacopeial standards.
We invite potential partners to engage with our technical procurement team to discuss how this optimized route can be integrated into your supply chain. By leveraging our expertise, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to request specific COA data and route feasibility assessments to verify how our implementation of this green chemistry approach can drive down your overall manufacturing costs while securing a reliable, high-quality supply of this vital pharmaceutical intermediate.