Advanced Synthesis of High-Purity Antiviral Intermediates for Commercial Pharmaceutical Production
The global pharmaceutical landscape has been profoundly reshaped by the urgent demand for effective antiviral therapeutics, particularly those targeting SARS-CoV-2. In this context, patent CN114591302B emerges as a critical technological breakthrough, offering a robust preparation method for compounds of formula (I) or their salts, which serve as key intermediates in the synthesis of potent 3CL protease inhibitors like S-217622. This patent addresses the longstanding challenges of low yield and insufficient purity that have plagued previous synthetic routes, providing a viable pathway for industrial mass production. The invention details a sophisticated chemical process that not only enhances the reaction efficiency but also ensures the final product meets the stringent quality standards required for pharmaceutical applications. By optimizing reaction conditions and purification steps, this technology enables the production of high-purity intermediates with superior physical properties, such as improved fluidity and anti-caking characteristics. For international pharmaceutical companies, this represents a significant opportunity to secure a reliable supply chain for next-generation antiviral medications. The technical depth of this patent underscores its value as a cornerstone for future drug development efforts against coronaviruses.
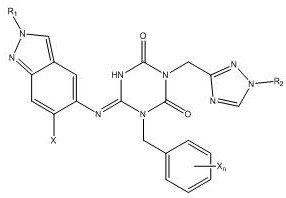
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations disclosed in CN114591302B, the synthesis of key antiviral intermediates faced severe bottlenecks that hindered large-scale commercialization. Existing literature, such as the work by Yuto Unoh et al., described methods that resulted in unacceptably low yields, often hovering around 25% in critical reaction steps. This inefficiency translates directly into excessive raw material consumption and heightened production costs, making the process economically unviable for mass manufacturing. Furthermore, the purity of the target product obtained through these conventional routes was typically limited to approximately 86%, which falls short of the rigorous specifications demanded by regulatory bodies for pharmaceutical ingredients. Low purity levels introduce complex impurity profiles that complicate downstream processing and pose risks to patient safety. Additionally, the physical properties of the crude products from older methods were often poor, exhibiting issues with fluidity and caking that disrupted formulation processes. These technical deficiencies created a significant barrier to entry for manufacturers aiming to produce cost-effective antiviral treatments at a global scale.
The Novel Approach
The novel approach presented in this patent fundamentally reengineers the synthesis pathway to overcome these historical limitations through precise control of reaction parameters and reagent addition. By introducing a specific protocol involving the reaction of a compound of formula (II) with a compound of formula (III) in the presence of a co-solvent and a base added in portions, the inventors have achieved a dramatic improvement in performance. This method consistently delivers yields exceeding 75%, representing a threefold increase over prior art, which drastically reduces the cost of goods sold per kilogram of active intermediate. Moreover, the purity of the resulting compound of formula (I) is routinely achieved at levels above 90%, with some embodiments reaching upwards of 97% after purification. This high level of chemical purity is complemented by enhanced physical characteristics, including excellent flowability and resistance to agglomeration, which are critical for automated tabletting and capsule filling operations. The strategic use of co-solvents and controlled temperature profiles ensures that the reaction proceeds smoothly without the formation of stubborn by-products, thereby simplifying the purification workflow and increasing overall throughput.
Mechanistic Insights into Base-Catalyzed Condensation Reaction
The core of this technological advancement lies in the meticulous optimization of the condensation reaction between the triazine-dione derivative and the indazol-amine precursor. The mechanism relies on the precise deprotonation of the active methylene or amine species using strong non-nucleophilic bases such as Lithium Hexamethyldisilazide (LHMDS) or potassium tert-butoxide. Unlike traditional single-addition methods, this patent advocates for a portion-wise addition of the base, initially at low temperatures ranging from 0°C to 5°C, followed by a gradual warming to room temperature or slightly higher. This controlled addition strategy minimizes exothermic spikes that can lead to decomposition or side reactions, ensuring a cleaner reaction profile. The presence of a co-solvent system, which may include mixtures of water with organic solvents like dioxane or acetonitrile, plays a pivotal role in solubilizing the reactants and stabilizing the transition states. This solvation effect facilitates the nucleophilic attack necessary for bond formation while maintaining the integrity of the sensitive functional groups present in the molecule. The result is a highly selective transformation that favors the formation of the desired imino-triazinane-dione structure over potential isomers or degradation products.
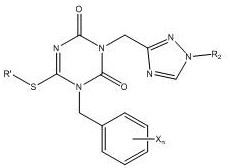
Impurity control is another critical aspect of this mechanism, achieved through the careful selection of reaction conditions that suppress the formation of known by-products. The patent highlights that maintaining the molar ratio of the base to the substrate within a specific range, typically 1:1 to 1:1.5, is essential for maximizing conversion while minimizing excess reagent waste. Furthermore, the use of anhydrous conditions or controlled water content in the co-solvent mixture prevents hydrolysis of the sensitive triazine ring, which is a common source of impurities in similar chemistries. Post-reaction, the purification strategy involving column chromatography or recrystallization from specific solvent pairs like acetone and water effectively removes residual starting materials and trace impurities. This multi-layered approach to impurity management ensures that the final product not only meets chemical purity standards but also possesses the physical homogeneity required for consistent dosing in pharmaceutical formulations. The robustness of this mechanism against variations in scale makes it particularly attractive for technology transfer from laboratory to pilot and commercial plants.
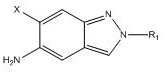
How to Synthesize High-Purity Antiviral Intermediate Efficiently
Implementing this synthesis route requires a disciplined approach to process engineering, focusing on the precise execution of the reaction steps outlined in the patent. The process begins with the preparation of the reaction vessel, ensuring it is dry and inert, followed by the charging of the compound of formula (II) and compound of formula (III) in a suitable solvent system such as tetrahydrofuran. The critical step involves the controlled addition of the base solution, which must be dosed slowly to maintain the reaction temperature within the specified low range initially. Detailed standard operating procedures for this synthesis, including specific stirring rates, addition times, and quenching protocols, are essential for reproducibility. The following guide summarizes the key operational phases required to achieve the high yields and purity levels described in the technical disclosure.
- Prepare the reaction system by dissolving compound of formula (II) and compound of formula (III) in a suitable organic solvent such as THF, ensuring the presence of a co-solvent to enhance solubility.
- Add a strong base such as LHMDS or potassium tert-butoxide to the reaction mixture in portions at controlled low temperatures (0-5°C) to initiate the condensation reaction.
- Maintain the reaction at elevated temperatures (20-30°C) after the initial addition, followed by quenching, extraction, and purification via recrystallization to achieve purity above 90%.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis method offers substantial strategic benefits that extend beyond mere technical specifications. The primary advantage lies in the significant reduction of manufacturing costs driven by the drastic improvement in reaction yield. By tripling the output from the same amount of raw materials, manufacturers can lower the unit cost of the intermediate, allowing for more competitive pricing in the final drug product or higher margins for the supplier. This efficiency gain also reduces the burden on waste management systems, as less unreacted material and by-products need to be treated and disposed of, contributing to a more sustainable and environmentally compliant operation. Furthermore, the enhanced purity and physical properties of the product streamline the downstream formulation process, reducing the risk of batch failures and production delays. This reliability translates into a more predictable supply chain, ensuring that pharmaceutical companies can meet market demand without the risk of shortages caused by manufacturing bottlenecks.
- Cost Reduction in Manufacturing: The elimination of inefficient reaction steps and the maximization of raw material utilization directly contribute to a leaner cost structure. By avoiding the need for extensive reprocessing or multiple purification cycles to achieve acceptable purity, the overall operational expenditure is significantly lowered. The use of commercially available solvents and reagents further ensures that the supply chain remains resilient against price volatility in specialty chemicals. This economic efficiency makes the production of complex antiviral intermediates more accessible and sustainable in the long term.
- Enhanced Supply Chain Reliability: The robustness of the new process ensures consistent batch-to-batch quality, which is critical for maintaining regulatory compliance and customer trust. High yield and purity reduce the likelihood of out-of-specification results that could halt production lines or delay shipments. This stability allows supply chain planners to optimize inventory levels and reduce safety stock requirements, freeing up working capital. Additionally, the scalability of the method means that production capacity can be ramped up quickly in response to surges in demand, such as during a pandemic outbreak, without compromising product quality.
- Scalability and Environmental Compliance: The process is designed with scale-up in mind, utilizing standard equipment and conditions that are easily transferable to large-scale reactors. The reduction in waste generation and the use of less hazardous solvent systems align with modern green chemistry principles, facilitating easier permitting and regulatory approval. This environmental stewardship not only mitigates regulatory risk but also enhances the corporate social responsibility profile of the manufacturing entity, appealing to eco-conscious partners and investors.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this novel synthesis technology. These answers are derived directly from the experimental data and technical disclosures within the patent documentation, providing clarity for stakeholders evaluating this process for adoption. Understanding these details is crucial for making informed decisions about integrating this method into existing production workflows or sourcing strategies.
Q: What is the primary advantage of the new synthesis method over prior art?
A: The new method significantly improves the yield from approximately 25% to over 75% and increases purity from 86% to above 90%, addressing critical industrial mass production limitations.
Q: How does the purity level affect the physical properties of the intermediate?
A: Achieving a purity of 90.0% or more results in significantly improved fluidity and anti-caking properties, which are essential for consistent tabletting and formulation development.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process utilizes common solvents and reagents with controlled temperature profiles, making it highly scalable and suitable for commercial production of antiviral intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable S-217622 Intermediate Supplier
As the global demand for effective antiviral therapies continues to evolve, partnering with a technically proficient CDMO is essential for bringing these critical medicines to market. NINGBO INNO PHARMCHEM stands at the forefront of this effort, leveraging deep expertise in complex organic synthesis to deliver high-quality pharmaceutical intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless. We are committed to meeting stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of S-217622 intermediate meets the highest standards of safety and efficacy required by international regulatory agencies.
We invite pharmaceutical partners to collaborate with us to optimize their supply chains and reduce time-to-market for vital treatments. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. We encourage you to reach out for specific COA data and route feasibility assessments to verify how our advanced manufacturing capabilities can support your drug development pipeline. Together, we can ensure a stable and efficient supply of life-saving medications for patients worldwide.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →