Industrial Scale-Up of Azvudine: A Robust 6-Step Synthetic Route for Global Supply
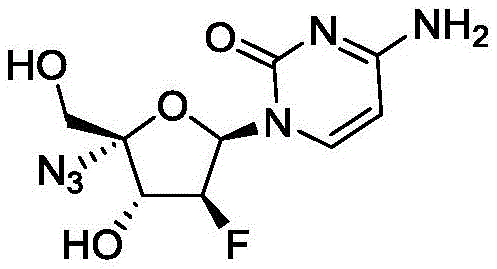
The pharmaceutical landscape for antiviral therapeutics has been significantly reshaped by the emergence of nucleoside analogs, particularly 2'-deoxy-2'-beta-fluoro-4'-azidocytidine, widely known as Azvudine or FNC. As detailed in the recent patent documentation CN114380877A, a breakthrough preparation method has been established that addresses the critical bottlenecks of scalability and safety inherent in earlier synthetic strategies. This innovative approach utilizes 2'-deoxy-2'-beta-fluoro-1,3,5-tribenzoyl-D-ribofuranose as the primary raw material, streamlining the production of this vital active pharmaceutical ingredient through a concise six-step reaction sequence. The significance of this development cannot be overstated for global supply chains, as FNC has demonstrated potent efficacy not only against HIV as a reverse transcriptase inhibitor but also shows promising clinical results in the treatment of novel coronavirus infections. By optimizing the synthetic pathway to avoid hazardous reagents and reduce step count, this technology offers a viable solution for the commercial scale-up of complex pharmaceutical intermediates, ensuring a more stable and cost-effective supply for downstream drug manufacturers.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this technological advancement, the synthesis of FNC was plagued by inefficiencies that rendered large-scale production economically and operationally challenging. The most credible existing routes, such as those developed by the Junbiao team, typically involved a cumbersome ten-step reaction sequence. A major drawback of these legacy methods was the reliance on high-risk explosive reagents, specifically sodium azide and iodine chloride, which pose severe safety hazards in an industrial setting and require specialized handling protocols that drive up operational costs. Furthermore, the purification burden in these traditional pathways was immense; nearly every intermediate step necessitated separation via column chromatography. This reliance on chromatographic purification is a significant bottleneck for manufacturing, as it limits batch sizes, increases solvent consumption drastically, and extends production lead times, ultimately resulting in a disappointing total yield ranging merely from 5.8% to 17.6%. Such low efficiency and high complexity make these conventional routes unsuitable for meeting the surging global demand for high-purity antiviral agents.
The Novel Approach
In stark contrast, the methodology disclosed in patent CN114380877A represents a paradigm shift towards lean and robust manufacturing. By condensing the synthesis into just six strategic steps, the new process drastically simplifies the operational workflow while simultaneously enhancing safety profiles. The route ingeniously avoids the aforementioned high-risk reagents, substituting them with safer, more manageable chemicals that do not compromise the stereochemical integrity of the final product. Crucially, the new method minimizes the need for labor-intensive column chromatography, favoring scalable unit operations such as crystallization, liquid-liquid extraction, and simple washing procedures. This transition from chromatographic dependence to crystallization-based purification is a key enabler for cost reduction in pharmaceutical intermediates manufacturing. The result is a process that not only achieves a superior total yield of 21% to 22% but also demonstrates strong operability, making it ideally suited for the rigorous demands of industrial-scale production where consistency and throughput are paramount.
Mechanistic Insights into Iron-Catalyzed Azidation and Oxidation
The core of this synthetic innovation lies in the sophisticated manipulation of the sugar moiety, particularly during the oxidation and azidation phases. The process employs Dess-Martin periodinane, a hypervalent iodine reagent, to selectively oxidize the hydroxyl group of the intermediate to a carbonyl functionality. This choice of oxidant is critical as it operates under mild conditions, preventing the degradation of sensitive functional groups elsewhere on the molecule, which is a common issue with harsher oxidizing agents. Following oxidation, the synthesis leverages a stereoselective azidation strategy mediated by an iron catalyst, such as ferrous chloride or ferric sulfate. This catalytic system facilitates the addition of the azido group and the subsequent ring closure with high fidelity. The use of an iron catalyst is mechanistically superior because it promotes the formation of the desired beta-configuration while suppressing side reactions that could lead to diastereomeric impurities. This precise control over stereochemistry is essential for ensuring the biological activity of the final nucleoside analog, as the wrong isomer could be inactive or even toxic.
Furthermore, the mechanism includes a carefully orchestrated deprotection and amination sequence in the final stages. The use of phosphorus oxychloride in conjunction with 1,2,4-triazole and pyridine allows for the efficient activation of the cytosine base for coupling, followed by a gentle deprotection using hydroxylamine and ammonia. This multi-step cascade is designed to maximize atom economy and minimize waste generation. From an impurity control perspective, the specific reaction conditions—such as maintaining temperatures between 0°C and 5°C during critical addition steps—ensure that exothermic reactions are managed effectively, preventing the formation of thermal degradation byproducts. The ability to monitor reaction progress via HPLC and stop reactions at precise conversion points (e.g., when raw material purity drops below 1%) ensures that the impurity profile remains tightly controlled throughout the synthesis. This level of mechanistic understanding and process control is what allows the production of high-purity pharmaceutical intermediates that meet the stringent regulatory standards required for API manufacturing.
How to Synthesize 2'-deoxy-2'-beta-fluoro-4'-azidocytidine Efficiently
The execution of this six-step synthesis requires precise adherence to the reaction parameters outlined in the patent to ensure optimal yield and purity. The process begins with the hydrolysis of the tribenzoyl starting material, followed by the critical oxidation and azidation steps that define the molecular architecture. Each stage, from the initial dissolution in dry organic solvents to the final crystallization of the white crystal compound FNC, is optimized for reproducibility. For research and development teams looking to implement this technology, the detailed standardized synthesis steps provided in the patent serve as a comprehensive guide for laboratory validation and pilot plant trials. The following section outlines the procedural framework necessary to achieve the reported efficiencies.
- Hydrolysis of the starting tribenzoyl ribofuranose using hydrogen bromide in acetic acid to form the intermediate hemiacetal.
- Oxidation of the hydroxyl group to a carbonyl using Dess-Martin periodinane under mild conditions.
- Stereoselective azidation and cyclization utilizing trimethylsilyl azide and an iron catalyst to construct the azido-sugar core.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route translates directly into tangible strategic advantages. The primary value driver is the significant simplification of the manufacturing process, which inherently lowers the cost of goods sold (COGS). By eliminating the need for multiple rounds of column chromatography, the process reduces solvent usage, labor hours, and equipment occupancy time, leading to substantial cost savings without the need for complex financial modeling. Additionally, the removal of high-risk explosive reagents like sodium azide simplifies regulatory compliance and reduces insurance and safety infrastructure costs, further enhancing the economic viability of the project. These factors combined create a more resilient supply chain capable of withstanding market fluctuations and raw material price volatility.
- Cost Reduction in Manufacturing: The transition from a ten-step to a six-step process fundamentally alters the cost structure of FNC production. Fewer reaction steps mean fewer unit operations, less energy consumption for heating and cooling, and a drastic reduction in the volume of organic solvents required for workups and purifications. The avoidance of expensive and hazardous reagents also contributes to a leaner bill of materials. Moreover, the higher total yield of 21-22% compared to the historical 5.8-17.6% means that less starting material is wasted, maximizing the output per batch and effectively lowering the unit cost of the final API. This efficiency gain is critical for maintaining competitive pricing in the global antiviral market.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by complex syntheses that are prone to failure at multiple stages. This robust six-step route mitigates such risks by utilizing stable intermediates and reliable reaction conditions. The ability to isolate intermediates via crystallization rather than chromatography ensures that the process can be easily scaled from kilogram to tonne quantities without losing efficiency. This scalability guarantees a consistent supply of high-purity pharmaceutical intermediates, reducing the risk of stockouts for downstream drug manufacturers. Furthermore, the use of commercially available and non-restricted reagents ensures that the supply chain is not vulnerable to geopolitical restrictions or specialized vendor dependencies.
- Scalability and Environmental Compliance: In the current regulatory environment, environmental sustainability is a key metric for supplier selection. This process aligns with green chemistry principles by minimizing waste generation and avoiding the use of heavy metals or highly toxic reagents where possible. The simplified workup procedures generate less hazardous waste, reducing disposal costs and environmental impact. The process is designed for easy scale-up, allowing manufacturers to rapidly increase production capacity to meet surges in demand, such as those seen during pandemic outbreaks. This agility, combined with a cleaner environmental footprint, positions suppliers adopting this technology as preferred partners for multinational pharmaceutical companies committed to sustainable sourcing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis method. These answers are derived directly from the technical specifications and beneficial effects described in the patent literature, providing clarity on the operational benefits and safety improvements of the new route. Understanding these details is crucial for stakeholders evaluating the feasibility of integrating this technology into their existing production portfolios.
Q: How does the new 6-step route compare to previous methods in terms of safety?
A: The new method eliminates the use of high-risk explosive reagents such as sodium azide and iodine chloride, replacing them with safer alternatives like trimethylsilyl azide and iron catalysts, significantly improving operational safety.
Q: What is the total yield improvement of this novel synthesis pathway?
A: While conventional routes offered total yields between 5.8% and 17.6%, this optimized 6-step process achieves a substantially higher total yield of approximately 21% to 22%.
Q: Does this process require extensive purification steps like column chromatography?
A: Unlike prior art which required column chromatography for most intermediates, this method relies primarily on crystallization, extraction, and washing, making it highly suitable for large-scale industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2'-deoxy-2'-beta-fluoro-4'-azidocytidine Supplier
The technical potential of this six-step synthesis route for 2'-deoxy-2'-beta-fluoro-4'-azidocytidine represents a significant opportunity for the pharmaceutical industry to secure a more stable and economical supply of this critical antiviral agent. At NINGBO INNO PHARMCHEM, we pride ourselves on our capability to translate such innovative patent technologies into commercial reality. As a seasoned CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this process are fully realized in practice. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of FNC intermediate meets the highest international standards for safety and efficacy.
We invite global pharmaceutical partners to collaborate with us to leverage this advanced manufacturing technology. By working together, we can optimize the supply chain and achieve meaningful Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you secure a reliable supply of high-quality nucleoside intermediates, ensuring your drug development and commercialization timelines are met with confidence and precision.