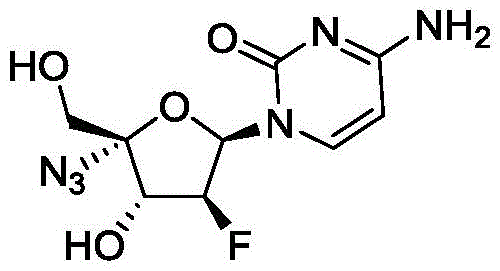
Industrial Scale-Up of 2'-Deoxy-2'-Beta-Fluoro-4'-Azidocytidine via Optimized Six-Step Catalysis
The pharmaceutical landscape for antiviral treatments has been significantly reshaped by the development of 2'-deoxy-2'-beta-fluoro-4'-azidocytidine, widely known as Azvudine or FNC. This potent nucleoside analogue serves as a critical reverse transcriptase inhibitor, demonstrating remarkable efficacy against HIV and showing promising results in the treatment of novel coronavirus infections. The technical breakthrough detailed in patent CN114380877B represents a paradigm shift in the manufacturing methodology for this high-value active pharmaceutical ingredient. By transitioning from cumbersome, low-yield legacy processes to a streamlined six-step catalytic route, the industry now has access to a preparation method that prioritizes both economic efficiency and operational safety. This report analyzes the profound implications of this patented technology for global supply chains, offering a roadmap for R&D directors and procurement leaders seeking to secure a reliable API supplier for next-generation antiviral therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of FNC has been plagued by inefficiencies that render large-scale production economically prohibitive and operationally hazardous. The pioneering work by the Chang Junbiao team, while scientifically valid, established a ten-step synthetic route that suffers from a critically low total yield ranging merely between 5.8% and 17.6%. A major bottleneck in this conventional approach is the heavy reliance on column chromatography for the purification of intermediates at nearly every stage, a technique that is notoriously difficult to scale beyond laboratory quantities due to excessive solvent consumption and time requirements. Furthermore, the traditional pathway necessitates the use of high-risk reagents such as sodium azide and iodine chloride, introducing severe safety liabilities regarding explosion hazards and toxic waste management. These factors collectively create a fragile supply chain where cost reduction in pharmaceutical manufacturing is stifled by poor atom economy and complex downstream processing requirements.
The Novel Approach
In stark contrast, the methodology disclosed in patent CN114380877B introduces a revolutionary six-step sequence that fundamentally重构 s the production logic for cost reduction in antiviral drug manufacturing. By strategically selecting 2'-deoxy-2'-beta-fluoro-1,3,5-tribenzoyl-D-ribofuranose as the starting material, the inventors have engineered a pathway that achieves a total yield of 21-22%, effectively doubling the output efficiency compared to prior art. The most significant operational improvement lies in the replacement of column chromatography with crystallization-based purification for key intermediates, a change that drastically simplifies the workflow and enhances reproducibility. Additionally, the novel route creatively avoids the use of dangerous inorganic azides, opting instead for safer organic azide sources catalyzed by iron salts. This shift not only mitigates safety risks but also streamlines the regulatory compliance process, making the commercial scale-up of complex pharmaceutical intermediates far more attainable for industrial partners.
Mechanistic Insights into Iron-Catalyzed Azidation and Oxidation
The core innovation of this synthesis lies in the sophisticated manipulation of stereochemistry during the ring-closing and azidation phases, specifically in Step 3. The process utilizes a Dess-Martin periodinane reagent to selectively oxidize the hydroxyl group of the intermediate to a carbonyl functionality, creating an electrophilic center primed for nucleophilic attack. Subsequently, the introduction of the azido group is mediated by an iron catalyst, such as ferrous chloride or ferric sulfate, in the presence of trimethylsilyl azide. This metal-catalyzed transformation is crucial because it ensures high stereoselectivity, favoring the formation of the desired beta-configuration while minimizing the generation of alpha-isomer impurities that are difficult to separate later. The mechanistic precision here allows the reaction to proceed under mild conditions, typically between 0°C and 40°C, preserving the integrity of the sensitive fluorine substituent which is essential for the drug's biological activity.
Impurity control is further reinforced through the strategic design of the protection and deprotection groups throughout the six-step cascade. The use of benzoyl groups in the early stages provides robust stability against harsh reaction conditions, while their removal in the final steps is carefully managed using ammonia methanol solutions to prevent degradation of the nucleobase. The patent specifies rigorous monitoring of liquid phase purity, ensuring that raw material conversion exceeds 98% before proceeding to subsequent steps. This attention to detail in the reaction mechanism translates directly to a cleaner crude product profile, reducing the burden on final purification stages. For R&D teams, understanding this mechanistic nuance is vital for troubleshooting potential batch variations and ensuring that the high-purity API intermediate specifications are consistently met during technology transfer.
How to Synthesize 2'-Deoxy-2'-Beta-Fluoro-4'-Azidocytidine Efficiently
Executing this synthesis requires strict adherence to the optimized reaction parameters outlined in the patent to maximize yield and safety. The process begins with the hydrolysis of the tribenzoyl starting material, followed by the critical oxidation and azidation steps that define the molecule's unique pharmacophore. Operators must maintain precise temperature controls, particularly during the exothermic addition of hydrogen bromide and the low-temperature phosphorylation in the final amination stage. The following guide summarizes the standardized operational protocol derived from the patent examples, serving as a foundational reference for process engineers preparing for pilot plant trials.
- Hydrolysis of the starting tribenzoyl ribofuranose using hydrogen bromide in acetic acid to form the dibenzoyl intermediate.
- Oxidation of the hydroxyl group to a carbonyl using Dess-Martin periodinane reagent to prepare for ring closure.
- Stereoselective azido group introduction and ring closure using an iron catalyst and trimethylsilyl azide.
- Bromination at the anomeric position followed by coupling with trimethylsilyl uracil to form the nucleoside base.
- Final amination and deprotection steps using phosphorus oxychloride, triazole, and ammonia methanol solution to yield FNC.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers transformative benefits that extend far beyond simple yield improvements. The elimination of column chromatography represents a massive reduction in solvent usage and processing time, directly translating to substantial cost savings in manufacturing overheads. By shortening the synthetic route from ten steps to six, the overall cycle time for production is drastically reduced, allowing for faster inventory turnover and improved responsiveness to market demand fluctuations. This efficiency gain is critical for maintaining a reliable supply of high-purity pharmaceutical intermediates in a volatile global market where lead times can often be a bottleneck for drug development programs.
- Cost Reduction in Manufacturing: The economic impact of this process is driven primarily by the removal of expensive and time-consuming purification steps. Traditional methods requiring repeated column chromatography consume vast quantities of silica gel and organic solvents, generating significant hazardous waste disposal costs. By shifting to crystallization-based purification, the new method minimizes material waste and energy consumption associated with solvent recovery. Furthermore, the avoidance of high-cost, high-risk reagents like sodium azide reduces the need for specialized safety infrastructure and insurance premiums, leading to a leaner and more cost-effective production model that enhances overall profit margins without compromising quality.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by the complexity of multi-step syntheses where a failure in any single step can halt the entire batch. The robustness of this six-step route, characterized by high conversion rates and simplified workup procedures, significantly lowers the risk of batch failures. The use of readily available starting materials and stable catalysts ensures that raw material sourcing remains consistent and unaffected by geopolitical or logistical disruptions. This reliability allows supply chain planners to forecast production schedules with greater accuracy, ensuring that downstream formulation teams receive their API allocations on time, thereby preventing costly delays in clinical trials or commercial product launches.
- Scalability and Environmental Compliance: Scaling chemical processes from the gram scale to multi-ton production often reveals hidden inefficiencies, but this route is explicitly designed for industrial operability. The replacement of chromatographic separation with crystallization is a key enabler for scalability, as crystallization tanks are standard equipment in any GMP facility and can be easily scaled up without linear increases in cost. Additionally, the reduced solvent load and the elimination of heavy metal contaminants or explosive azide salts simplify the environmental compliance landscape. This makes the process more sustainable and easier to permit in regions with strict environmental regulations, securing the long-term viability of the manufacturing site.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and beneficial effects reported in patent CN114380877B, providing clarity on yield expectations, safety protocols, and scalability potential. Understanding these details is essential for stakeholders evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: How does the new 6-step route improve upon previous 10-step methods?
A: The new method described in patent CN114380877B reduces the synthetic sequence from 10 steps to just 6 steps, significantly increasing the total yield from approximately 5.8-17.6% to over 21-22%. Furthermore, it eliminates the need for column chromatography in multiple intermediate steps, replacing them with crystallization, which is far more viable for industrial scale-up.
Q: What safety advantages does this process offer regarding azide reagents?
A: Unlike conventional routes that rely on hazardous sodium azide and iodine chloride, this novel approach utilizes safer azide sources such as trimethylsilyl azide or diphenyl phosphorazidate in conjunction with iron catalysts. This substitution drastically reduces the explosion risks associated with handling inorganic azides in large-scale manufacturing environments.
Q: Is this synthesis route suitable for multi-ton commercial production?
A: Yes, the process is specifically designed for industrial operability. By avoiding complex purification techniques like column chromatography and utilizing robust crystallization methods for intermediates, the route ensures high throughput and consistent quality control, making it ideal for reliable pharmaceutical intermediate suppliers aiming for commercial volume.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2'-Deoxy-2'-Beta-Fluoro-4'-Azidocytidine Supplier
As the global demand for effective antiviral therapies continues to surge, the ability to produce high-quality 2'-deoxy-2'-beta-fluoro-4'-azidocytidine at scale has become a strategic priority for pharmaceutical companies worldwide. NINGBO INNO PHARMCHEM stands at the forefront of this challenge, leveraging our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production to bring this patented technology to life. Our state-of-the-art facilities are equipped to handle the specific requirements of fluorinated nucleoside synthesis, ensuring that every batch meets stringent purity specifications through our rigorous QC labs. We understand that consistency is key in API manufacturing, and our dedicated technical team is committed to delivering a supply of FNC that supports your critical drug development timelines without compromise.
We invite you to collaborate with us to unlock the full commercial potential of this efficient synthesis route. Whether you are looking to optimize an existing supply chain or initiate a new project, our experts are ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team today to request specific COA data and comprehensive route feasibility assessments. Together, we can ensure a stable, cost-effective, and high-quality supply of this vital antiviral intermediate, driving success for your organization and improving patient outcomes globally.