Advanced Six-Step Synthesis of Brexpiprazole Intermediates: Enhancing Selectivity and Commercial Scalability
Advanced Six-Step Synthesis of Brexpiprazole Intermediates: Enhancing Selectivity and Commercial Scalability
The pharmaceutical industry constantly seeks robust, scalable, and cost-effective pathways for the synthesis of complex active pharmaceutical ingredients (APIs) and their critical intermediates. A significant breakthrough in this domain is documented in patent CN114249720A, which details a novel preparation method for Brexpiprazole, a potent atypical antipsychotic agent. This technology addresses long-standing challenges in the synthesis of key intermediates such as 3-[4-[4-(benzo[b]thiophen-4-yl)piperazin-1-yl]butoxy]aniline. By integrating a streamlined six-step sequence that includes alcoholysis, nucleophilic substitution, catalytic reduction, and advanced palladium-catalyzed couplings, this method offers a superior alternative to legacy processes. For R&D directors and supply chain leaders, understanding the mechanistic advantages and operational efficiencies of this route is crucial for securing a reliable supply of high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Brexpiprazole intermediates has been plagued by issues related to selectivity, safety, and operational complexity. Prior art, such as the methods described in WO2018172463A1, often relies on the use of 1-bromo-4-chlorobutane for alcoholysis. However, the close activity of halogen atoms at both ends of this molecule leads to poor reaction selectivity, resulting in a mixture of isomers and byproducts that are difficult to separate. Furthermore, traditional routes frequently employ Boc (tert-butyloxycarbonyl) groups to protect amino functionalities. While effective in theory, the Boc group is unstable under the alkaline conditions required for subsequent alkylation steps, leading to premature deprotection and the generation of complex impurities. Additionally, some existing patents utilize electrochemical reduction methods which, while chemically interesting, pose significant hurdles for commercial scale-up due to equipment constraints and the risk of reducing sensitive double bonds on the thiophene ring, creating structural analogs that are notoriously difficult to remove.
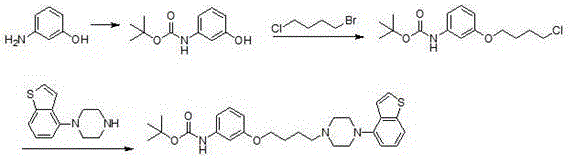
The Novel Approach
In stark contrast to these cumbersome legacy methods, the novel approach outlined in CN114249720A introduces a highly efficient six-step synthetic strategy that circumvents these pitfalls. The core innovation lies in the strategic selection of starting materials and reaction conditions that maximize selectivity while minimizing waste. Instead of struggling with unstable protecting groups, this method utilizes a direct alcoholysis of 3-nitrophenol with specific iodohydrocarbons, followed by a seamless transition to piperazine substitution. Crucially, the process avoids the use of hydroxyquinolinone and eliminates the need for electrochemical reduction, thereby removing the risk of over-reduction on the thiophene moiety. The route is designed such that the first three steps—alcoholysis, substitution, and reduction—can be executed with continuous feeding. This operational modification not only reduces the complexity of handling intermediate isolations but also drives the overall yield of this critical sequence to exceed 60.0 percent, a significant improvement over batch-wise legacy processes.
Mechanistic Insights into Palladium-Catalyzed Cyclization and Coupling
The heart of this synthetic advancement lies in its sophisticated use of transition metal catalysis, specifically palladium-mediated transformations. The conversion of the aniline intermediate (Compound IV) to the benzo[b]thiophene-substituted piperazine (Compound V) is achieved via a Buchwald-Hartwig amination. In this step, a palladium catalyst, such as tetrakis(triphenylphosphine)palladium, facilitates the cross-coupling between the aryl chloride of 4-chlorobenzo[b]thiophene and the secondary amine of the piperazine ring. The presence of a strong base like sodium tert-butoxide is critical for generating the reactive amido-palladium species. This mechanism ensures high regioselectivity, attaching the bulky heterocyclic system precisely at the desired nitrogen atom without affecting other sensitive functional groups on the butoxy chain. Following this, the synthesis proceeds through an acylation with cinnamoyl chloride derivatives to form an amide linkage, setting the stage for the final ring closure.
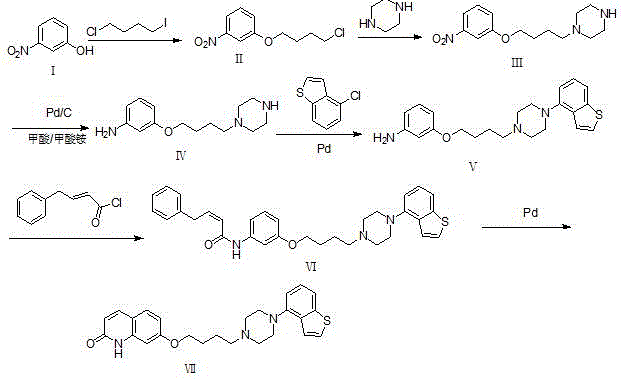
The final and most critical transformation is the intramolecular palladium-catalyzed cyclization of Compound VI to yield the target Brexpiprazole (Compound VII). This step involves the formation of a new carbon-carbon or carbon-heteroatom bond to close the quinolinone ring system. The patent specifies the use of specialized palladium catalysts, such as tetrakis(tri-o-methylphenylphosphine)palladium, at elevated temperatures (110-115 ℃). The choice of ligand and catalyst loading (3-5% w) is optimized to ensure complete conversion while allowing for the recovery and reuse of the precious metal. This recyclability is a key mechanistic feature that translates directly into economic value. By preventing the formation of polymeric side products and ensuring clean cyclization, the process maintains a high purity profile, with related substances controlled below 0.5 percent in optimized examples, demonstrating exceptional impurity control mechanisms inherent to the catalytic cycle.
How to Synthesize Brexpiprazole Intermediates Efficiently
Implementing this novel synthesis requires precise control over reaction parameters to fully realize its benefits. The process begins with the alcoholysis of 3-nitrophenol using 1-chloro-4-iodobutane in methanol with potassium hydroxide, maintaining a temperature of 20-30 ℃ to prevent side reactions. The resulting chloro-ether is then reacted with piperazine in a polar aprotic solvent like DMF at 80-85 ℃. The subsequent reduction of the nitro group is performed using formic acid and ammonium formate as a hydrogen source with a palladium on carbon catalyst at 50-55 ℃. These initial steps set the foundation for the high-yield continuous feeding strategy. For the detailed standardized operating procedures, safety protocols, and specific workup instructions required for GMP manufacturing, please refer to the technical guide below.
- Perform alcoholysis of 3-nitrophenol with 1-chloro-4-iodobutane to form the nitro-ether intermediate, followed by immediate piperazine substitution.
- Reduce the nitro group to an amine using a palladium catalyst and formic acid/ammonium formate hydrogen source.
- Execute a palladium-catalyzed Buchwald-Hartwig coupling with 4-chlorobenzo[b]thiophene, followed by acylation and final intramolecular cyclization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthesis route offers compelling strategic advantages beyond mere chemical elegance. The primary benefit is the drastic simplification of the supply chain for raw materials. By avoiding exotic or highly regulated precursors like hydroxyquinolinone and replacing them with commodity chemicals such as 3-nitrophenol and standard iodohydrocarbons, the process mitigates the risk of raw material shortages. Furthermore, the elimination of electrochemical steps removes the need for specialized, hard-to-source reactor equipment, allowing production to be scaled in standard stainless steel vessels found in most multipurpose API plants. This flexibility significantly enhances supply continuity and reduces the lead time for scaling up production capacities to meet market demand.
- Cost Reduction in Manufacturing: The economic impact of this method is profound, driven primarily by the efficient use of catalysts and reagents. The patent explicitly highlights that the palladium catalysts used in the coupling and cyclization steps can be recycled and reused. In traditional processes where precious metals are lost in aqueous waste streams, this recyclability represents a direct and substantial saving in raw material costs. Additionally, the ability to continuously feed reagents in the first three steps reduces solvent consumption and labor hours associated with multiple isolation and purification cycles. The higher overall yield (>60.0% for the first three steps) means less starting material is wasted, further driving down the cost of goods sold (COGS) for the final API intermediate.
- Enhanced Supply Chain Reliability: Reliability in the pharmaceutical supply chain is often compromised by complex purification requirements and low yields. This novel route addresses these issues by improving reaction selectivity, which inherently reduces the burden on downstream purification units. With fewer byproducts and impurities generated, the reliance on extensive chromatographic purification is minimized, leading to faster batch turnover times. The robustness of the chemistry, which tolerates standard industrial conditions without requiring cryogenic temperatures or high-pressure hydrogenation equipment, ensures that production schedules are less likely to be disrupted by technical failures or equipment maintenance, providing a more predictable delivery timeline for downstream drug product manufacturers.
- Scalability and Environmental Compliance: From an environmental and regulatory perspective, this method aligns perfectly with modern green chemistry principles. The avoidance of Lewis acids in the final cyclization step eliminates the generation of large volumes of acidic aluminum or iron-containing waste, which is costly and difficult to treat. Similarly, bypassing electrochemical reduction removes the energy intensity and safety risks associated with high-current processes. The use of recyclable catalysts and the reduction of solvent-intensive purification steps contribute to a lower E-factor (mass of waste per mass of product). This not only simplifies compliance with increasingly stringent environmental regulations but also positions the manufacturer as a sustainable partner, a key criterion for many global pharmaceutical buyers today.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Brexpiprazole synthesis technology. These insights are derived directly from the comparative data and experimental results presented in the patent literature, offering a clear view of the process capabilities.
Q: How does this novel method improve upon traditional Brexpiprazole synthesis routes?
A: Unlike conventional methods that rely on unstable Boc protection groups or complex electrochemical reductions, this novel route utilizes a continuous feeding strategy for the first three steps. This approach significantly improves reaction selectivity, avoids the generation of harmful benzene byproducts, and achieves a cumulative yield exceeding 60.0% for the initial sequence, thereby simplifying post-treatment and purification.
Q: What are the cost advantages regarding catalyst usage in this process?
A: The process employs palladium catalysts in key coupling and reduction steps which can be effectively recycled and reused. This recyclability drastically reduces the consumption of expensive precious metals compared to single-use catalytic systems, leading to substantial raw material cost optimization without compromising reaction efficiency or product purity.
Q: Is this synthesis route suitable for large-scale commercial production?
A: Yes, the method is specifically designed for scalability. By avoiding electrochemical reduction steps which are difficult to scale and eliminating the need for harsh Lewis acids that complicate waste treatment, the process offers a robust pathway for industrial manufacturing. The ability to continuously feed reagents in the early stages further enhances operational efficiency and throughput potential.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Brexpiprazole Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent laboratory data to commercial reality requires deep technical expertise and robust infrastructure. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facilities are equipped to handle the specific requirements of palladium-catalyzed reactions, including dedicated recovery systems for precious metals and rigorous QC labs capable of detecting trace impurities to meet stringent purity specifications. We understand the critical nature of CNS active ingredients and are committed to delivering Brexpiprazole intermediates that consistently meet the highest quality standards required by global regulatory bodies.
We invite you to leverage our technical capabilities to optimize your supply chain. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team to request specific COA data, route feasibility assessments, and samples for your qualification process. Let us collaborate to bring this advanced, cost-effective synthesis technology to your commercial production lines, ensuring a secure and efficient supply of this vital pharmaceutical intermediate.