Advanced Repaglinide Manufacturing: Optimized Amidation and Deprotection Strategies for Commercial Scale
Advanced Repaglinide Manufacturing: Optimized Amidation and Deprotection Strategies for Commercial Scale
The pharmaceutical industry continuously seeks robust synthetic routes for high-volume antidiabetic agents, and patent CN102786498A presents a significant advancement in the preparation of Repaglinide. This novel methodology addresses critical bottlenecks in traditional synthesis, specifically focusing on the optimization of the amidation coupling and the subsequent deprotection sequences. By shifting away from toxic condensing agents and implementing a precise temperature-controlled hydrolysis protocol, this process offers a pathway to higher purity and improved safety profiles. For R&D directors and process chemists, understanding these mechanistic nuances is vital for evaluating the feasibility of technology transfer. The following analysis dissects the technical merits of this approach, highlighting its potential to redefine supply chain standards for this essential meglitinide class medication.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
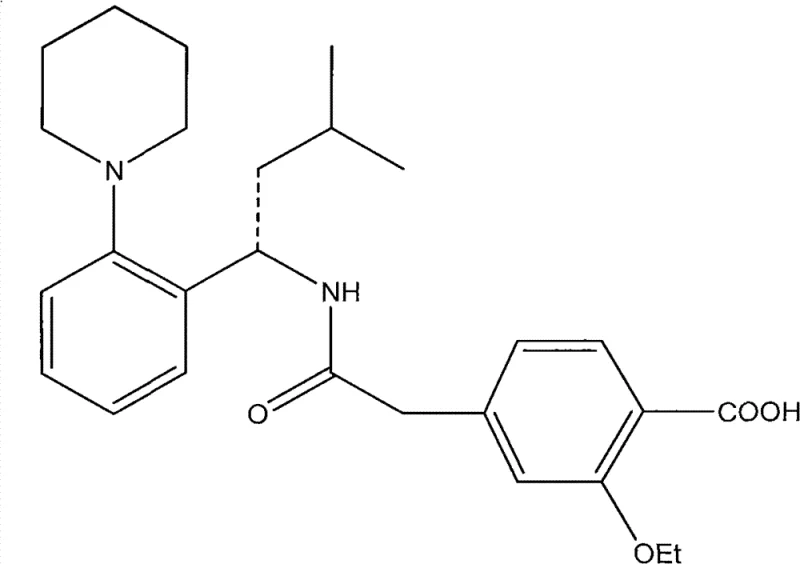
Historically, the synthesis of Repaglinide has relied heavily on condensation strategies described in earlier literature, such as US Patent 5312924. These conventional routes typically involve the direct coupling of the chiral amine fragment with the benzoic acid derivative using reagents like DCC (dicyclolhexylcarbodiimide) or triphenylphosphine combined with carbon tetrachloride. While effective on a laboratory scale, these reagents introduce severe drawbacks for industrial application. The use of DCC generates dicyclohexylurea (DCU) as a byproduct, which is notoriously difficult to remove completely from the reaction mixture, often requiring complex filtration and recrystallization steps that lower overall throughput. Furthermore, reagents like carbon tetrachloride pose significant environmental and occupational health hazards, complicating waste disposal and regulatory compliance. The reaction times associated with these older methods are also frequently prolonged, leading to lower space-time yields in manufacturing reactors.
The Novel Approach
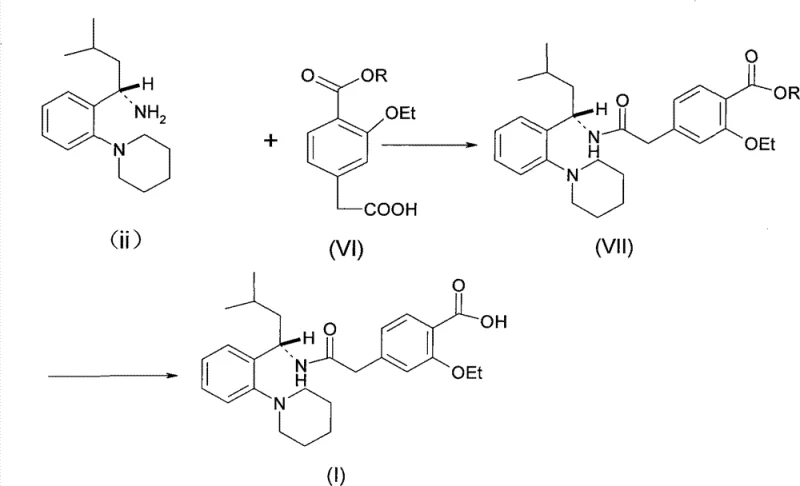
In contrast, the methodology disclosed in CN102786498A introduces a refined synthetic strategy that mitigates these risks through the use of superior coupling agents and a sequential deprotection protocol. The core innovation lies in the utilization of CDI (carbonyldiimidazole) or similar activated ester forming agents, which facilitate cleaner amidation reactions without generating insoluble urea precipitates. More critically, the patent outlines a specific order of operations for removing protecting groups: the carboxyl protecting group (R2) is removed first under alkaline conditions, followed by the removal of the amine protecting group (R1) under controlled acidic conditions. This sequential approach prevents the potential for side reactions that can occur when both groups are exposed to harsh conditions simultaneously. By optimizing the acidolysis temperature to a narrow window of 30-40°C, the process achieves a remarkable balance between reaction rate and product stability, resulting in significantly higher yields and purity compared to the prior art.
Mechanistic Insights into CDI-Mediated Amidation and Sequential Hydrolysis
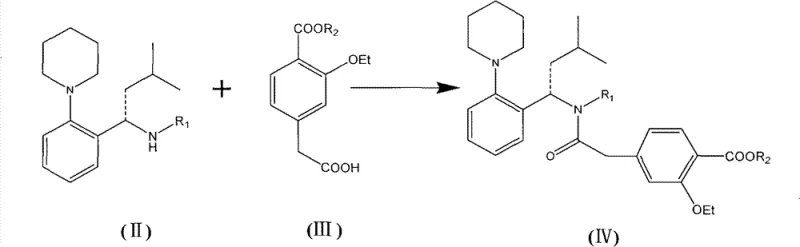
The chemical efficacy of this new route is rooted in the enhanced nucleophilicity of the amine component and the chemoselectivity of the deprotection steps. In the initial amidation phase, the introduction of the R1 group (specifically a p-methoxybenzyl or similar moiety) on the amine nitrogen serves a dual purpose: it protects the amine from unwanted oxidation or side reactions during storage and handling, and electronically activates the nitrogen center, thereby increasing its nucleophilicity towards the activated carboxyl species generated by CDI. This electronic activation allows the coupling reaction to proceed more rapidly and at milder temperatures, reducing the energy input required for the process. The resulting intermediate (Formula IV) is stable enough to be isolated or carried forward without significant degradation, providing flexibility in the manufacturing schedule.
Furthermore, the mechanism of the sequential deprotection is a masterclass in impurity control. Traditional methods often struggle with the simultaneous removal of ester and amine protecting groups, which can lead to racemization at the chiral center or the formation of polymeric byproducts. By first subjecting the intermediate to alkaline hydrolysis, the ester moiety (R2) is selectively cleaved to reveal the free carboxylic acid while the amine protection remains intact. This step is followed by a carefully controlled acidolysis using a mixture of trifluoroacetic acid and methanesulfonic acid. The patent data indicates that maintaining the temperature strictly between 30°C and 40°C during this acid step is crucial; temperatures outside this range can lead to incomplete deprotection or the degradation of the sensitive peptide-like bond. This precise control ensures that the final Repaglinide product retains its stereochemical integrity and meets stringent pharmacopeial standards for enantiomeric excess.
How to Synthesize Repaglinide Efficiently
The execution of this synthesis requires strict adherence to the optimized parameters defined in the patent to ensure reproducibility and high yield. The process begins with the preparation of the chiral amine intermediate, followed by the key coupling reaction with the protected benzoic acid derivative. Operators must pay close attention to the stoichiometry of the coupling agent and the temperature profile during the workup phases. The subsequent hydrolysis steps demand precise pH control and temperature monitoring to avoid the formation of difficult-to-remove impurities. For a detailed breakdown of the specific reagent quantities, solvent systems, and operational timelines required to execute this synthesis successfully, please refer to the standardized protocol outlined below.
- Condense the chiral amine intermediate (Formula II) with the protected benzoic acid derivative (Formula III) using CDI as the coupling agent to form the protected amide (Formula IV).
- Perform alkaline hydrolysis to selectively remove the carboxyl protecting group (R2) while maintaining the amine protection.
- Execute acidic hydrolysis using a trifluoroacetic acid and methanesulfonic acid mixture at 30-40°C to remove the amine protecting group (R1) and isolate the final Repaglinide product.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this optimized synthesis route offers substantial benefits for procurement managers and supply chain directors looking to secure a reliable repaglinide intermediate supplier. The shift away from hazardous reagents like carbon tetrachloride and the elimination of difficult-to-filter byproducts like DCU directly translates to a safer and more efficient manufacturing environment. This not only reduces the operational costs associated with waste treatment and safety compliance but also minimizes the risk of production stoppages due to equipment fouling or regulatory audits. The simplified purification process means that fewer unit operations are required to achieve the desired purity, effectively shortening the overall production cycle time and enhancing the responsiveness of the supply chain to market demands.
- Cost Reduction in Manufacturing: The replacement of expensive and toxic condensing agents with CDI, combined with the higher yields achieved through optimized deprotection, leads to a significant reduction in the cost of goods sold. By eliminating the need for extensive purification steps to remove urea byproducts, the process consumes less solvent and energy, driving down variable production costs. Additionally, the improved yield means that less raw material is wasted per kilogram of final product, maximizing the return on investment for precursor chemicals and ensuring a more economically viable production model for high-volume API manufacturing.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route contributes to greater supply chain stability. Because the reaction conditions are milder and less prone to runaway exotherms or sensitivity to moisture compared to older methods, the risk of batch failure is drastically reduced. This reliability allows for more accurate forecasting and inventory planning, ensuring that downstream formulation teams receive their materials on schedule. The use of commercially available starting materials and standard reagents further mitigates the risk of supply disruptions, making this a highly dependable source for critical diabetes medication intermediates.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, with temperature controls and reagent choices that are easily managed in large-scale reactors. The reduction in toxic waste streams aligns with modern green chemistry principles, facilitating easier permitting and environmental compliance in diverse global jurisdictions. This scalability ensures that the manufacturing capacity can be expanded from pilot plant levels to multi-ton commercial production without the need for fundamental process re-engineering, providing a seamless path for meeting growing global demand for antidiabetic therapies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These insights are derived directly from the experimental data and process descriptions found in the patent documentation, providing a clear understanding of the operational advantages. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this route for their own manufacturing portfolios or for procurement specialists assessing supplier capabilities.
Q: Why is CDI preferred over DCC for Repaglinide synthesis?
A: CDI (Carbonyldiimidazole) is preferred because it avoids the formation of insoluble dicyclohexylurea (DCU) byproducts associated with DCC, simplifying purification and reducing toxicity risks in large-scale API manufacturing.
Q: What is the critical temperature range for the final acidolysis step?
A: The patent specifies a critical temperature range of 30-40°C for the acid hydrolysis step. Maintaining this range is essential to maximize yield and minimize the formation of thermal degradation impurities.
Q: How does the sequential deprotection strategy improve purity?
A: By removing the ester group (R2) under alkaline conditions first, followed by the amine protecting group (R1) under acidic conditions, the process prevents side reactions and ensures a more complete conversion compared to simultaneous deprotection methods.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Repaglinide Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and compliant synthesis routes for life-saving medications like Repaglinide. Our technical team has extensively analyzed the methodologies presented in CN102786498A and possesses the expertise to implement these advanced coupling and deprotection strategies at an industrial scale. We boast extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent supply of high-quality intermediates. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch meets the exacting standards required for pharmaceutical applications.
We invite potential partners to engage with our technical procurement team to discuss how this optimized process can benefit your specific supply chain needs. By leveraging our manufacturing capabilities, you can access a Customized Cost-Saving Analysis tailored to your volume requirements. We encourage you to contact us today to request specific COA data and route feasibility assessments, allowing you to make informed decisions that drive efficiency and profitability in your antidiabetic drug portfolio.