Advanced Manufacturing of Tenofovir: A Novel Synthetic Route for High-Purity API Production
The pharmaceutical industry continuously seeks robust manufacturing pathways for critical antiviral agents, particularly for treating chronic conditions like Hepatitis B and HIV. Patent CN101906119B discloses a significant advancement in the preparation of Tenofovir, specifically its monohydrate form, which serves as a cornerstone in modern antiretroviral therapy. This intellectual property outlines a novel synthetic methodology that diverges from traditional, cumbersome routes by employing a strategic trityl protection scheme on (R)-1,2-propanediol. The disclosed process addresses long-standing challenges in nucleoside phosphonate synthesis, including the reliance on hazardous reducing agents and complex purification sequences. By streamlining the reaction pathway, this technology offers a viable solution for manufacturers aiming to enhance production efficiency while maintaining stringent quality standards required for active pharmaceutical ingredients.
For R&D directors and process chemists, the implications of this patent extend beyond mere novelty; it represents a tangible opportunity to optimize the impurity profile and overall yield of Tenofovir production. The method leverages well-understood organic transformations, such as nucleophilic substitution and acid-catalyzed hydrolysis, but arranges them in a sequence that minimizes side reactions and maximizes atom economy. As global demand for generic antivirals continues to rise, adopting such efficient synthetic routes becomes a competitive necessity for any serious reliable pharmaceutical intermediates supplier looking to secure market share through superior process chemistry.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Tenofovir has been plagued by inefficiencies inherent in early-generation routes, often characterized by excessive step counts and the use of specialized reagents. As illustrated in prior art, conventional methods frequently initiate with D-(+)-isobutyl lactate, necessitating a protection step with dihydropyrane followed by reduction using diisobutylaluminum hydride or similar complex hydrides. These reagents are not only costly but also pose significant safety hazards on a large industrial scale due to their pyrophoric nature and sensitivity to moisture. Furthermore, these legacy routes typically involve multiple cycles of protection and deprotection to manage the reactivity of hydroxyl groups, leading to substantial material loss at each stage.
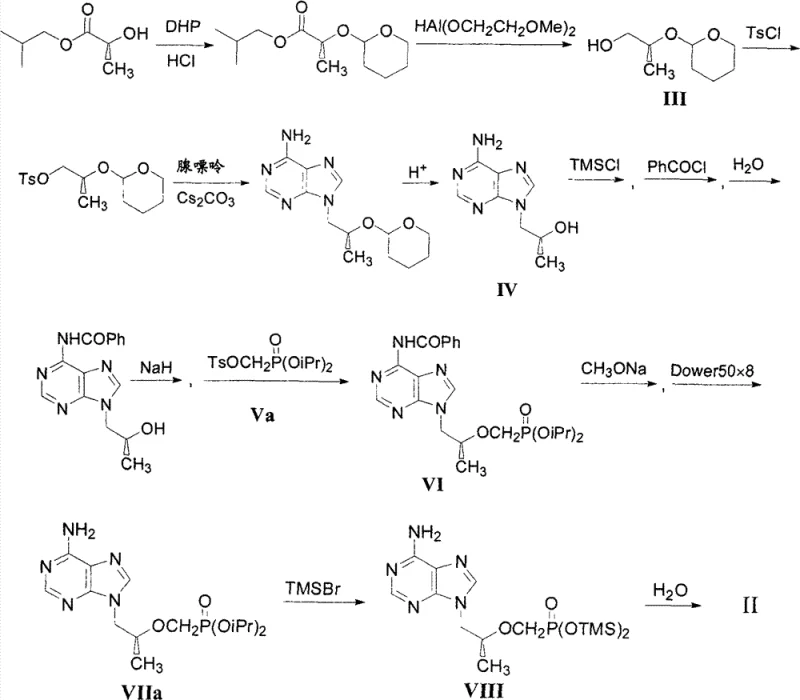
Another critical drawback of existing bibliographic methods, such as those relying on (S)-glycidol derivatives, is the instability of optical purity. Documents indicate that while some routes appear brief, they often yield products with suboptimal enantiomeric excess, necessitating additional chirality enrichment steps. These enrichment processes are notoriously unpredictable, with yields fluctuating significantly, which introduces unacceptable variability into the supply chain. The cumulative effect of these limitations is a manufacturing process that is difficult to scale, expensive to operate, and prone to generating complex impurity profiles that complicate downstream purification and regulatory approval.
The Novel Approach
In stark contrast to the convoluted pathways of the past, the novel method described in patent CN101906119B introduces a streamlined approach centered on the selective protection of (R)-1,2-propanediol. By utilizing substituted triphenylchloromethane (trityl chloride) to protect the terminal hydroxyl group, the process effectively differentiates the two hydroxyl functionalities without the need for cyclic acetals or harsh reducing conditions. This strategic move allows for the direct condensation of the remaining secondary hydroxyl group with dialkyl sulfonyloxy methyl phosphonate, establishing the crucial carbon-phosphorus bond early in the sequence with high fidelity.
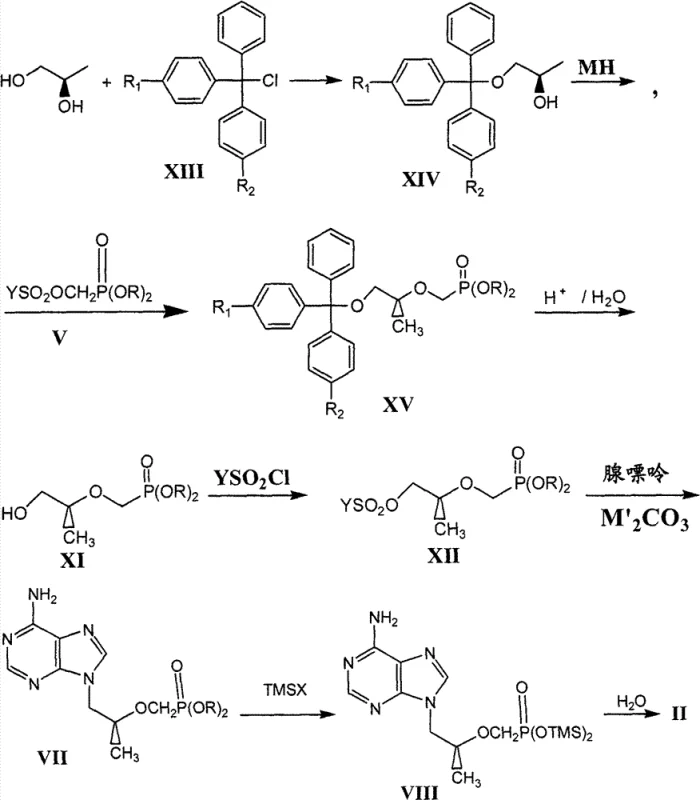
The elegance of this new route lies in its operational simplicity and the accessibility of its reagents. Following the initial coupling, the trityl protecting group is removed under mild organic acid catalysis, a transformation that generates triphenylcarbinol as a byproduct which can be easily separated via filtration or extraction. This ease of purification stands in sharp relief to the chromatographic separations often required in older methods. Subsequent conversion to the sulfonate and coupling with adenine proceeds under standard conditions, ultimately yielding the target phosphonate ester which is then hydrolyzed to the final acid. This logical progression reduces the overall number of unit operations, directly translating to cost reduction in pharmaceutical intermediates manufacturing and a smaller environmental footprint.
Mechanistic Insights into Trityl-Mediated Phosphonate Coupling
The core mechanistic advantage of this synthesis rests on the steric bulk and acid-lability of the trityl group. In the initial step, the reaction between (R)-1,2-propanediol and triphenylmethyl chloride is driven by the preferential formation of the primary trityl ether. The bulky trityl cation selectively attacks the less hindered primary hydroxyl group, leaving the secondary hydroxyl available for subsequent functionalization. This regioselectivity is paramount, as it obviates the need for temporary protecting groups on the secondary alcohol, thereby shortening the synthetic tree. The use of organic bases like triethylamine serves to scavenge the generated hydrogen chloride, driving the equilibrium forward without introducing metallic salts that could complicate waste streams.
Following protection, the generation of the alkoxide using an alkali metal hydride, such as sodium hydride, creates a potent nucleophile at the secondary position. This species attacks the methylene carbon of the dialkyl sulfonyloxy methyl phosphonate in an SN2-type displacement. The sulfonyloxy group acts as an excellent leaving group, facilitating the formation of the ether linkage that connects the propyl chain to the phosphonate moiety. Crucially, the reaction conditions are tuned to prevent elimination side reactions, preserving the integrity of the carbon backbone. The subsequent hydrolysis step exploits the unique stability profile of the trityl ether; while stable to base, it is rapidly cleaved by weak organic acids like acetic acid. This orthogonality allows for the selective deprotection of the primary alcohol in the presence of the phosphonate ester and the newly formed ether linkage, setting the stage for the final coupling with the adenine base.
How to Synthesize Tenofovir Efficiently
The implementation of this novel synthetic route requires precise control over reaction parameters to ensure optimal yield and purity. The process begins with the careful addition of trityl chloride to the diol substrate, maintaining temperature control to manage the exotherm. Subsequent steps involving strong bases like sodium hydride demand strictly anhydrous conditions to prevent reagent decomposition. The condensation with adenine, a key step in forming the nucleoside analog, requires elevated temperatures in polar aprotic solvents to overcome the low nucleophilicity of the purine base. Detailed standard operating procedures for each transformation, including specific molar ratios, solvent choices, and workup protocols, are essential for successful technology transfer.
- Protect the terminal hydroxyl group of (R)-1,2-propanediol using substituted triphenylchloromethane to form the trityl ether intermediate.
- React the protected alcohol with an alkali metal hydride to form the alkoxide, followed by condensation with dialkyl sulfonyloxy methyl phosphonate.
- Hydrolyze the trityl protecting group under organic acid catalysis to reveal the primary hydroxyl group.
- Convert the free hydroxyl group into a sulfonate leaving group using sulfonyl chloride.
- Perform nucleophilic substitution with adenine in the presence of a mineral base to attach the purine base.
- Transform the phosphonate ester into the final phosphonic acid using trimethylsilyl bromide followed by aqueous hydrolysis.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the shift away from specialized reducing agents and chiral catalysts towards commodity chemicals represents a significant strategic win. The primary starting material, (R)-1,2-propanediol, is widely available from multiple global suppliers, mitigating the risk of single-source dependency. Similarly, reagents such as triphenylmethyl chloride and adenine are produced at massive scales for various industries, ensuring price stability and consistent quality. This transition to a robust, commodity-based supply chain enhances the resilience of the manufacturing operation against market fluctuations and logistical disruptions.
- Cost Reduction in Manufacturing: The elimination of expensive and hazardous reducing agents, such as diisobutylaluminum hydride, drastically lowers the raw material cost per kilogram of product. Furthermore, the reduction in the number of synthetic steps directly correlates with lower utility consumption, reduced labor hours, and decreased solvent usage. The ability to isolate intermediates through simple crystallization or filtration, rather than resource-intensive column chromatography, further drives down the cost of goods sold, making the final API more competitive in the generic marketplace.
- Enhanced Supply Chain Reliability: By removing dependencies on niche reagents that may have long lead times or limited production capacity, this method secures the continuity of supply. The use of standard organic solvents like dichloromethane, THF, and acetonitrile ensures that solvent procurement remains straightforward. Additionally, the robustness of the trityl protection strategy means that the process is less sensitive to minor variations in raw material quality, reducing the frequency of batch failures and ensuring a steady flow of high-purity pharmaceutical intermediates to downstream customers.
- Scalability and Environmental Compliance: The simplified workflow facilitates easier scale-up from pilot plant to commercial production volumes. Fewer unit operations mean less equipment turnover and reduced potential for cross-contamination. From an environmental standpoint, the avoidance of heavy metal catalysts and the generation of easily manageable byproducts like triphenylcarbinol simplify waste treatment protocols. This alignment with green chemistry principles not only reduces disposal costs but also supports corporate sustainability goals, a critical factor for modern commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel Tenofovir synthesis method. These insights are derived directly from the comparative data and experimental examples provided in the patent literature, offering clarity on process feasibility and performance metrics.
Q: How does this novel method improve upon conventional Tenofovir synthesis routes?
A: Unlike conventional routes that rely on expensive reducing agents like diisobutylaluminum hydride and multiple protection-deprotection cycles, this method utilizes a robust trityl protection strategy on readily available (R)-1,2-propanediol. This significantly shortens the synthetic sequence, eliminates the need for specialized reducing reagents, and avoids unstable chirality enrichment steps, resulting in a more stable and scalable process.
Q: What are the key advantages regarding raw material availability for this process?
A: The process relies on commodity chemicals such as (R)-1,2-propanediol, triphenylmethyl chloride, and adenine. By avoiding proprietary or highly specialized chiral catalysts and rare reducing agents found in prior art, the supply chain risk is minimized, ensuring consistent availability for large-scale commercial production.
Q: How is optical purity maintained throughout the synthesis?
A: The synthesis begins with enantiomerically pure (R)-1,2-propanediol. The reaction conditions, particularly the mild acid hydrolysis for deprotection and the specific alkylation steps, are designed to preserve the stereochemical integrity of the chiral center without requiring post-synthesis recrystallization or chiral resolution, thereby maintaining high optical purity throughout the workflow.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tenofovir Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes in the modern pharmaceutical landscape. Our technical team has thoroughly analyzed the methodology disclosed in CN101906119B and possesses the expertise to adapt this novel trityl-protection strategy for industrial-scale production. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to manufacturing plant is seamless. Our facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of Tenofovir intermediate meets the highest international standards.
We invite procurement leaders and R&D heads to collaborate with us on optimizing their supply chains for antiviral medications. By leveraging our process development capabilities, we can provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team to request specific COA data and route feasibility assessments, allowing you to make informed decisions that drive value and security into your API supply chain.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →