Advanced Synthesis of Fluvastatin Intermediates: A Technical Breakthrough for Commercial API Production
The pharmaceutical industry continuously seeks robust synthetic routes for HMG-CoA reductase inhibitors, commonly known as statins, due to their critical role in managing cardiovascular health. Patent CN1687032A introduces a transformative approach to synthesizing Fluvastatin intermediates, specifically addressing the longstanding challenge of stereochemical control in the dihydroxyheptanoic acid side chain. This technology shifts the synthetic paradigm by utilizing a novel 3-hydroxy-5-oxo heptanoate intermediate (Formula III), which fundamentally alters the reduction dynamics compared to traditional 5-hydroxy-3-oxo precursors. For R&D directors and process chemists, this represents a significant leap forward, offering a pathway that eliminates the need for cumbersome stereoisomer separation steps that have historically plagued statin manufacturing. The ability to directly produce active pharmaceutical ingredient (API) ready intermediates with a cis/trans ratio exceeding 99.5:0.5 without chromatographic purification is a compelling value proposition for any organization aiming to streamline their supply chain for cardiovascular medications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Fluvastatin and its analogues relied heavily on 5-hydroxy-3-oxo heptanoate derivatives as the key precursors for the dihydroxy side chain. As documented in prior art such as EP-A-0363934, achieving the requisite cis-stereochemistry (3R,5S/3S,5R) using these traditional substrates presented severe industrial hurdles. When using methyl esters, which are preferred for cost and handling reasons in large-scale production, the stereoselectivity was often insufficient, yielding trans-isomer impurities greater than 1 percent. To mitigate this, manufacturers were forced to utilize bulky tert-butyl esters, which improved selectivity but introduced significant downstream processing burdens, particularly the difficult removal of tert-butanol byproducts during the final saponification step. Furthermore, the reliance on these suboptimal substrates often necessitated energy-intensive recrystallization or chromatographic separation to meet the strict pharmacopeial limits for trans-isomers, which are capped at 0.8 percent. These inefficiencies not only inflated manufacturing costs but also extended lead times and increased solvent consumption, creating ecological and economic liabilities for producers.
The Novel Approach
The innovative methodology disclosed in CN1687032A circumvents these legacy issues by reversing the oxidation state of the side chain, employing a 3-hydroxy-5-oxo heptanoate structure (Formula III) as the reduction substrate. This structural inversion allows for a highly stereoselective reduction that favors the desired cis-diol configuration inherently, rather than relying on thermodynamic equilibrium or bulky protecting groups to force selectivity. By constructing the carbon skeleton via a Horner-Wadsworth-Emmons coupling between a specialized phosphonate (Formula IV) and the indole aldehyde, the process establishes the necessary framework with high E-selectivity before the critical reduction step. 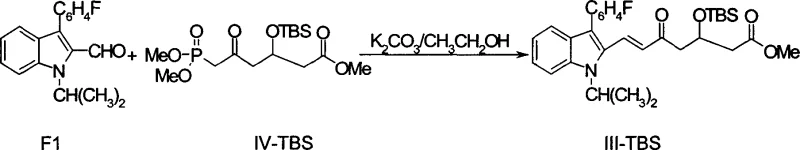 . This strategic placement of the hydroxyl group at the 3-position, protected typically as a TBS ether, directs the subsequent borane-mediated reduction of the 5-ketone with exceptional fidelity. Consequently, even when using industrially advantageous methyl esters, the process achieves a cis/trans ratio of up to 99.8 percent, rendering the expensive and time-consuming isomer separation steps entirely obsolete and significantly simplifying the overall manufacturing workflow.
. This strategic placement of the hydroxyl group at the 3-position, protected typically as a TBS ether, directs the subsequent borane-mediated reduction of the 5-ketone with exceptional fidelity. Consequently, even when using industrially advantageous methyl esters, the process achieves a cis/trans ratio of up to 99.8 percent, rendering the expensive and time-consuming isomer separation steps entirely obsolete and significantly simplifying the overall manufacturing workflow.
Mechanistic Insights into Stereoselective Borane-Mediated Reduction
The cornerstone of this synthetic advancement lies in the mechanistic nuances of the ketone reduction step, where the spatial arrangement of the 3-hydroxy group plays a pivotal directing role. The process utilizes an alkoxydialkylborane species, specifically methoxydiethylborane, which is generated in situ by reacting triethylborane with methanol under air catalysis. This Lewis acidic borane species coordinates with the carbonyl oxygen of the 5-ketone and the adjacent 3-hydroxyl group, forming a rigid cyclic transition state that locks the conformation of the molecule.  . Within this chelated complex, the hydride delivery from sodium borohydride is sterically guided to attack the carbonyl carbon from the less hindered face, ensuring the formation of the 5-hydroxyl group in the correct stereochemical relationship to the existing 3-hydroxyl group. This intramolecular directing effect is far superior to intermolecular reduction methods, as it minimizes the formation of the undesired trans-isomer. The reaction is typically conducted in a mixture of tetrahydrofuran and methanol at cryogenic temperatures, ranging from -78°C to -80°C, to further suppress non-selective background reduction and maximize the kinetic control over the stereochemical outcome.
. Within this chelated complex, the hydride delivery from sodium borohydride is sterically guided to attack the carbonyl carbon from the less hindered face, ensuring the formation of the 5-hydroxyl group in the correct stereochemical relationship to the existing 3-hydroxyl group. This intramolecular directing effect is far superior to intermolecular reduction methods, as it minimizes the formation of the undesired trans-isomer. The reaction is typically conducted in a mixture of tetrahydrofuran and methanol at cryogenic temperatures, ranging from -78°C to -80°C, to further suppress non-selective background reduction and maximize the kinetic control over the stereochemical outcome.
From an impurity control perspective, this mechanism offers a robust defense against the formation of diastereomeric byproducts that are notoriously difficult to separate in statin chemistry. In conventional routes, the lack of a proximal directing group often leads to a mixture of syn and anti diols, requiring multiple purification cycles that degrade overall yield. In contrast, the chelation-controlled reduction described here ensures that the primary reaction product is already of sufficient purity to proceed directly to the final saponification step. The use of ammonium bifluoride for the deprotection of the silyl ether prior to or during the reduction sequence is also critical, as it cleanly removes the protecting group without affecting the sensitive beta-hydroxy ketone motif or the indole ring system. This precise control over the reaction environment and reagent selection demonstrates a deep understanding of physical organic chemistry, translating directly into a cleaner crude product profile that reduces the burden on downstream purification units and analytical quality control laboratories.
How to Synthesize Fluvastatin Intermediate Efficiently
The practical execution of this synthesis involves a logical sequence of chain elongation, coupling, and selective functional group manipulation that is amenable to kilogram-scale production. The process begins with the preparation of the phosphonate side chain, where 3-protected glutaric anhydride undergoes alcoholysis to form a monoester, followed by phosphorylation using dimethyl methylphosphonate and n-butyllithium to install the Horner-Wadsworth-Emmons reagent. This phosphonate is then coupled with the indole aldehyde under basic conditions, typically using potassium carbonate in ethanol, to forge the carbon-carbon double bond with high E-selectivity. Following the coupling, the silyl protecting group is removed, and the resulting 3-hydroxy-5-oxo ester is subjected to the critical borane reduction. The detailed standardized synthesis steps, including specific molar ratios, temperature profiles, and workup procedures optimized for safety and yield, are provided in the structured guide below.
- Prepare the phosphonate side chain (IV-TBS) via alcoholysis of glutaric anhydride followed by phosphorylation and esterification.
- Perform Horner-Wadsworth-Emmons coupling between the phosphonate (IV-TBS) and the indole aldehyde (F1) to form the protected ketone (III-TBS).
- Deprotect the silyl group and perform stereoselective reduction using methoxydiethylborane and sodium borohydride to yield the cis-diol intermediate.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain executives, the adoption of this synthetic route offers tangible benefits that extend beyond mere technical elegance, directly impacting the bottom line through operational efficiency and risk mitigation. The elimination of stereoisomer separation steps, such as preparative chromatography or repeated recrystallizations, translates to a drastic reduction in solvent consumption and waste disposal costs, which are major cost drivers in API manufacturing. Furthermore, the ability to use methyl esters instead of tert-butyl esters simplifies the raw material portfolio and avoids the logistical challenges associated with removing volatile tert-butanol byproducts, thereby enhancing the overall throughput of the production facility. This streamlined process flow reduces the number of unit operations required, which in turn shortens the manufacturing cycle time and improves the responsiveness of the supply chain to market demand fluctuations for cardiovascular therapeutics.
- Cost Reduction in Manufacturing: The primary economic advantage stems from the significant simplification of the purification train. By achieving high stereoselectivity directly in the reaction vessel, the need for expensive resin-based chromatography or multiple solvent-intensive recrystallization steps is removed. This not only saves on the direct cost of solvents and stationary phases but also reduces the labor and energy costs associated with running these additional processes. Additionally, the use of commodity reagents like triethylborane and sodium borohydride, rather than exotic chiral catalysts, keeps the raw material costs predictable and low, ensuring a stable cost of goods sold (COGS) structure that is resilient to market volatility.
- Enhanced Supply Chain Reliability: The reliance on robust, well-understood chemical transformations enhances the reliability of the supply chain. The reagents required for this synthesis, such as dimethyl methylphosphonate and indole aldehydes, are commercially available from multiple global suppliers, reducing the risk of single-source bottlenecks. Moreover, the process tolerance is high, meaning that minor variations in raw material quality do not catastrophically impact the yield or purity, providing a buffer against supply chain disruptions. The simplified workflow also means that production can be scaled up more rapidly in response to sudden increases in demand, ensuring continuity of supply for downstream drug product manufacturers who rely on timely delivery of high-quality intermediates.
- Scalability and Environmental Compliance: From an environmental and regulatory standpoint, this process aligns well with green chemistry principles by minimizing waste generation and solvent usage. The avoidance of heavy metal catalysts or toxic chromium-based oxidants reduces the environmental footprint of the manufacturing site and simplifies the regulatory filing process regarding residual impurities. The scalability of the Horner-Wadsworth-Emmons reaction and the borane reduction is well-documented, allowing for seamless transfer from pilot plant to multi-ton commercial production without the need for extensive re-optimization. This ease of scale-up ensures that the technology can meet the growing global demand for statins while maintaining compliance with increasingly stringent environmental regulations regarding effluent discharge and solvent emissions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route for Fluvastatin intermediates. These answers are derived directly from the experimental data and technical disclosures within the patent documentation, providing a reliable basis for feasibility assessments. Understanding these details is crucial for technical teams evaluating the integration of this chemistry into their existing manufacturing platforms or for procurement specialists negotiating supply agreements for these critical building blocks.
Q: How does this new route improve stereoselectivity compared to conventional methods?
A: By utilizing a 3-hydroxy-5-oxo intermediate instead of the traditional 5-hydroxy-3-oxo structure, the new method achieves a cis/trans ratio of up to 99.8:0.2 without requiring difficult chromatographic separation.
Q: Why is the methyl ester preferred over the tert-butyl ester in this process?
A: While tert-butyl esters offered better selectivity in older methods, they create significant waste removal issues during saponification. This new route allows the use of industrially friendly methyl esters while maintaining high stereochemical purity.
Q: What are the critical reagents for the stereoselective reduction step?
A: The process relies on the in-situ generation of methoxydiethylborane from triethylborane and methanol under air catalysis, followed by reduction with sodium borohydride at low temperatures (-78°C).
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fluvastatin Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-purity intermediates in the production of life-saving cardiovascular medications. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated stereochemical control required for Fluvastatin synthesis is maintained at every stage of manufacturing. We operate stringent purity specifications and utilize rigorous QC labs equipped with advanced chiral HPLC capabilities to guarantee that every batch of intermediate meets or exceeds the 99.5 percent cis-isomer purity benchmark established by this innovative technology. Our commitment to quality assurance means that our clients can rely on a consistent supply of material that facilitates smooth downstream processing and final API registration.
We invite pharmaceutical partners to engage with our technical procurement team to discuss how this advanced synthetic route can be tailored to your specific production needs. By leveraging our expertise in process optimization, we can provide a Customized Cost-Saving Analysis that quantifies the potential efficiencies for your specific operation. We encourage you to contact us to request specific COA data and route feasibility assessments, allowing you to make informed decisions that enhance your supply chain resilience and competitive positioning in the global statin market.