Scalable Synthesis of Tricyclic Aminoalcohol Intermediates via Advanced Asymmetric Reduction Technology
The pharmaceutical industry continuously seeks robust and cost-effective pathways for synthesizing complex active pharmaceutical ingredient (API) intermediates, particularly those required for treating metabolic disorders such as diabetes and obesity. Patent CN1360574A discloses a groundbreaking methodology for the preparation of tricyclic aminoalcohol derivatives of general formula (1), which serve as critical precursors in the development of therapeutic agents for these conditions. This intellectual property represents a significant leap forward in process chemistry by addressing the longstanding challenges associated with stereochemical control and purification efficiency. Unlike traditional approaches that often rely on cumbersome resolution steps or hazardous reagents, this novel process leverages advanced asymmetric catalysis to establish chirality early in the synthetic sequence. The strategic design of this pathway ensures that key intermediates possess favorable physical properties, such as high crystallinity, which drastically simplifies downstream processing. For R&D directors and procurement specialists evaluating supply chain resilience, understanding the mechanistic underpinnings and operational advantages of this technology is essential for securing a reliable source of high-purity pharmaceutical intermediates.
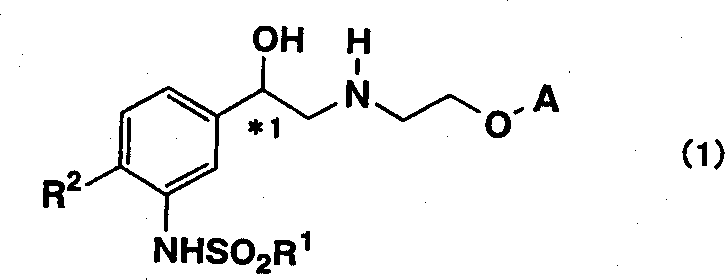
The limitations of conventional methods for synthesizing tricyclic aminoalcohol derivatives have historically posed significant barriers to efficient commercial manufacturing. Prior art, such as the methods disclosed in WO97/25311, often necessitates the use of expensive chiral auxiliaries, specifically borane-based reducing agents, to achieve the desired optical purity. These conventional routes are not only cost-prohibitive due to the high price of the chiral reagents but also introduce substantial safety hazards owing to the flammable and reactive nature of boranes. Furthermore, the resulting intermediates in these older processes frequently lack crystallinity, forcing manufacturers to rely on resource-intensive purification techniques like column chromatography, which are impractical for large-scale production. The requirement for strict anhydrous conditions and complex post-treatment procedures further exacerbates the operational costs and extends the production lead time, making these legacy methods less attractive for modern, high-volume pharmaceutical supply chains.
In stark contrast, the novel approach detailed in CN1360574A offers a streamlined and industrially viable alternative that circumvents these critical bottlenecks. The core innovation lies in the generation of new, crystalline intermediates, specifically the chlorohydrin compounds of formula (5) and the epoxide compounds of formula (4), which can be effectively purified through simple recrystallization rather than chromatography. This shift from chromatographic purification to crystallization-based purification represents a paradigm change in process economics, significantly reducing solvent consumption and waste generation. By establishing the stereocenter via asymmetric reduction of a chloroketone precursor using a transition metal catalyst, the process achieves high optical purity without the need for stoichiometric chiral auxiliaries. This methodology not only enhances the overall yield but also improves the safety profile of the manufacturing process by eliminating dangerous reagents, thereby providing a more sustainable and cost-efficient route for the production of these valuable pharmaceutical intermediates.
Mechanistic Insights into Ruthenium-Catalyzed Asymmetric Transfer Hydrogenation
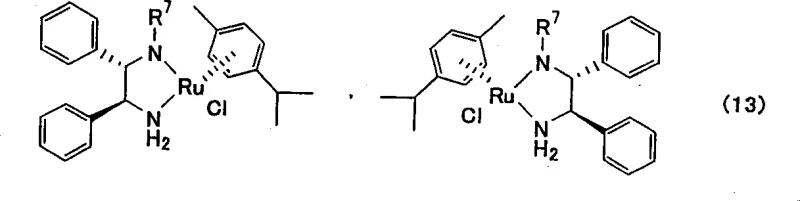
The heart of this innovative synthetic strategy is the asymmetric reduction step, which transforms the prochiral alpha-chloro ketone into the chiral chlorohydrin with exceptional enantioselectivity. This transformation is mediated by sophisticated ruthenium complexes, typically featuring chiral ethylenediamine ligands such as [(S,S)-N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine](p-cymene)ruthenium. The catalytic cycle operates through a concerted outer-sphere mechanism where the metal-ligand bifunctionality plays a pivotal role in activating both the hydride source and the ketone substrate. The chiral environment created by the diamine ligand dictates the facial selectivity of the hydride attack, ensuring the preferential formation of the desired R-configuration at the asymmetric carbon atom. This catalytic system is remarkably robust, functioning effectively under mild conditions with hydrogen donors like formic acid or isopropanol, which avoids the need for high-pressure hydrogenation equipment. The ability to tune the ligand structure allows for precise optimization of the reaction rate and stereoselectivity, making it a versatile tool for generating chiral building blocks.
Impurity control is another critical aspect where this mechanistic approach excels, directly impacting the quality of the final API intermediate. The formation of crystalline intermediates acts as a powerful purge point for impurities, including minor stereoisomers and side products generated during the reduction or subsequent cyclization steps. When the chlorohydrin or epoxide intermediates are subjected to recrystallization, the crystal lattice selectively incorporates the major enantiomer while excluding impurities, thereby upgrading the optical purity without additional chemical transformations. This phenomenon is particularly advantageous in the context of regulatory compliance, as it ensures a consistent and well-defined impurity profile for the final drug substance. Furthermore, the stability of these intermediates allows for isolation and storage, providing flexibility in the manufacturing schedule and enabling quality control testing before proceeding to the next synthetic stage. This level of control over the impurity spectrum is essential for meeting the stringent specifications required by global health authorities for pharmaceutical ingredients.
How to Synthesize Tricyclic Aminoalcohol Efficiently
The synthesis of these complex molecules involves a sequence of well-defined chemical transformations that prioritize yield and purity at every stage. The process begins with the chlorination of an acetophenone derivative to generate an alpha-chloro ketone, which serves as the substrate for the critical asymmetric reduction. Following the establishment of chirality, the chlorohydrin is cyclized to an epoxide using a base, creating a highly reactive electrophile ready for nucleophilic attack. The final assembly of the tricyclic structure is achieved by opening the epoxide ring with a functionalized amine, followed by the removal of protecting groups to reveal the active pharmacophore. Each step has been optimized to minimize side reactions and maximize the recovery of material, ensuring that the overall process is economically viable.
- Chlorination of acetophenone derivative followed by asymmetric reduction using a ruthenium catalyst to form chiral chlorohydrin.
- Cyclization of the chlorohydrin to an epoxide intermediate which can be purified via recrystallization.
- Ring-opening of the epoxide with a protected amine followed by deprotection to yield the final tricyclic aminoalcohol.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route translates into tangible strategic benefits that extend beyond mere technical feasibility. The elimination of expensive chiral borane auxiliaries and the replacement of chromatographic purification with crystallization significantly lowers the cost of goods sold (COGS). This cost reduction in pharmaceutical intermediate manufacturing is driven by the decreased consumption of high-value reagents and the reduced volume of solvents required for purification, which also lowers waste disposal costs. Moreover, the use of robust transition metal catalysts, which can often be recovered or used in low loadings, further contributes to the economic efficiency of the process. These factors combined create a more competitive pricing structure for the final intermediate, allowing pharmaceutical companies to optimize their budget allocation for drug development programs.
Enhanced supply chain reliability is another major advantage conferred by this technology, primarily due to the stability and isolability of the key intermediates. The ability to produce and store crystalline intermediates like the epoxide and chlorohydrin decouples the production stages, mitigating the risk of batch failures propagating through the entire synthesis. This modularity allows for better inventory management and ensures a continuous supply of materials even if one step encounters temporary delays. Additionally, the avoidance of hazardous reagents simplifies logistics and storage requirements, reducing the regulatory burden associated with transporting dangerous chemicals. By implementing a process that relies on standard unit operations such as filtration and crystallization, manufacturers can leverage existing infrastructure without the need for specialized equipment, thereby reducing lead time for high-purity pharmaceutical intermediates and accelerating time-to-market for new therapies.
Scalability and environmental compliance are inherently addressed by the design of this synthetic pathway, making it suitable for commercial scale-up of complex pharmaceutical intermediates. The transition from batch processes involving chromatography to continuous or semi-continuous crystallization processes aligns with green chemistry principles by minimizing solvent usage and energy consumption. The improved atom economy of the catalytic asymmetric reduction compared to stoichiometric chiral reductions reduces the generation of chemical waste, facilitating easier compliance with increasingly strict environmental regulations. Furthermore, the robustness of the reaction conditions allows for safe operation at larger scales, reducing the risk of thermal runaways or other safety incidents. This combination of scalability, safety, and environmental stewardship makes the technology an attractive option for contract development and manufacturing organizations (CDMOs) looking to expand their portfolio of sustainable manufacturing capabilities.
Frequently Asked Questions (FAQ)
The following questions address common inquiries regarding the technical and commercial implications of this patented synthesis method. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the practical application of the technology. Understanding these details helps stakeholders make informed decisions about integrating this pathway into their supply chains.
Q: What is the key advantage of the new synthesis method over conventional routes?
A: The new method eliminates the need for expensive chiral borane auxiliaries and complex chromatography, utilizing crystalline intermediates that allow for purification via simple recrystallization.
Q: Which catalyst is preferred for the asymmetric reduction step?
A: Ruthenium complexes, specifically those containing chiral ethylenediamine ligands such as [(S,S)-N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine](p-cymene)ruthenium, are highly effective.
Q: Can this process be scaled for commercial production?
A: Yes, the formation of crystalline intermediates like the chlorohydrin and epoxide facilitates easy handling and purification, making the process highly suitable for industrial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tricyclic Aminoalcohol Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of accessing high-quality intermediates for the successful development of metabolic disease treatments. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial manufacturing is seamless. We are committed to delivering products that meet stringent purity specifications through our rigorous QC labs, utilizing advanced analytical techniques to verify the identity and optical purity of every batch. Our facility is equipped to handle the specific requirements of asymmetric synthesis, including the safe handling of transition metal catalysts and the execution of precise crystallization protocols necessary for this technology.
We invite you to collaborate with us to leverage this advanced synthetic route for your next project. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this process can optimize your budget. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise in process chemistry can support your supply chain goals. By partnering with NINGBO INNO PHARMCHEM, you gain access to a reliable partner dedicated to driving innovation and efficiency in the pharmaceutical industry.