Advanced Solvent-Free Synthesis of Pregabalin Impurity II for Global Quality Control Standards
Advanced Solvent-Free Synthesis of Pregabalin Impurity II for Global Quality Control Standards
The pharmaceutical industry faces increasing regulatory pressure to identify and quantify trace impurities in Active Pharmaceutical Ingredients (APIs) to ensure patient safety. Patent CN113804805A introduces a groundbreaking methodology for the preparation of Pregabalin intermediate impurity compound II, a critical reference standard required for the quality control of Pregabalin. This innovation addresses the significant challenge of sourcing high-purity impurity standards by utilizing a novel solvent-free and catalyst-free synthetic route. By leveraging the reactant phenylethylamine as both a reagent and a solvent, the process eliminates the need for volatile organic compounds (VOCs) during the reaction phase, aligning perfectly with modern green chemistry principles. The resulting compound exhibits exceptional purity levels exceeding 99%, making it an ideal candidate for use as an impurity reference substance in rigorous drug quality control protocols.
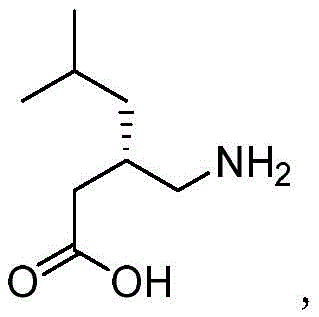
In the complex landscape of pharmaceutical manufacturing, the control of impurities is not merely a regulatory checkbox but a fundamental aspect of drug safety and efficacy. Pregabalin, a gamma-aminobutyric acid (GABA) analogue used for treating neuropathic pain and epilepsy, requires meticulous monitoring of its synthetic pathway. The presence of specific byproducts can alter the pharmacological profile or introduce toxicity. Therefore, the ability to synthesize these specific impurities in high purity is paramount for developing robust analytical methods. This patent provides a reliable solution for generating the necessary reference materials that allow manufacturers to detect and limit these unwanted byproducts effectively.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the acquisition of specific impurity standards like Compound II has been fraught with difficulties. In standard Pregabalin synthesis, Compound II is formed as an unavoidable byproduct when residual R-phenylethylamine reacts with urea during the production of the key intermediate, Compound III. Isolating this impurity from the reaction mother liquor is often inefficient, yielding low quantities with insufficient purity for analytical certification. Furthermore, conventional synthetic routes for urea derivatives frequently rely on hazardous solvents and transition metal catalysts, which introduce additional purification burdens to remove trace metals and solvent residues. These traditional methods often result in variable yields and inconsistent purity profiles, complicating the validation of analytical methods required by regulatory bodies like the FDA or EMA.
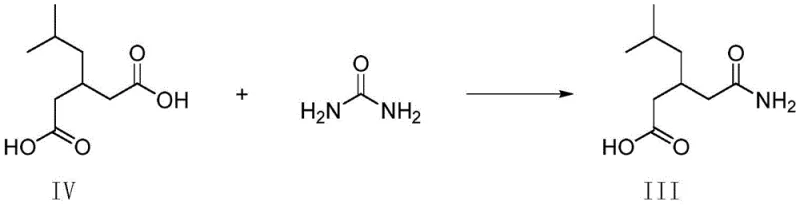
The Novel Approach
The methodology disclosed in patent CN113804805A represents a paradigm shift in how reference standards are manufactured. By reacting phenylethylamine directly with urea under thermal conditions without any added solvent or catalyst, the process achieves a streamlined synthesis with remarkable efficiency. The reaction utilizes the liquid phenylethylamine itself as the reaction medium, creating a homogeneous system that facilitates molecular collision and reaction progress. This approach not only simplifies the operational procedure but also drastically reduces the environmental footprint by eliminating solvent waste streams. The subsequent purification via simple recrystallization yields a product with purity greater than 99%, providing a robust and reproducible source of high-quality impurity standards for the global supply chain.
Mechanistic Insights into Thermal Condensation of Urea Derivatives
The core chemical transformation involves a thermal condensation reaction between a primary amine (phenylethylamine) and urea. Under elevated temperatures ranging from 90°C to 150°C, the nucleophilic nitrogen of the amine attacks the carbonyl carbon of the urea molecule. This attack leads to the displacement of an ammonia molecule, resulting in the formation of the substituted urea linkage found in Compound II. The absence of a catalyst necessitates precise temperature control to overcome the activation energy barrier, typically optimized between 110°C and 120°C. This thermal driving force ensures complete conversion while minimizing the formation of secondary degradation products that might arise from harsher acidic or basic catalytic conditions. The stoichiometry is carefully managed, with a molar ratio of phenylethylamine to urea maintained between 1:2.0 and 1:3 to drive the equilibrium towards the product side.
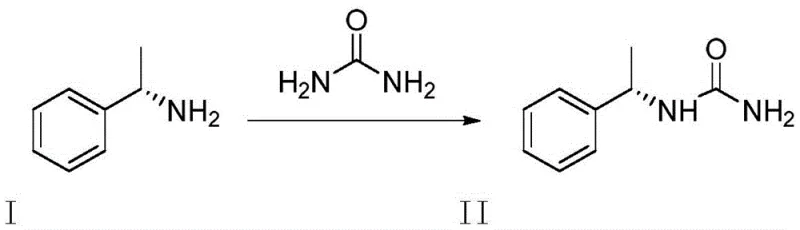
Controlling the impurity profile in this synthesis is achieved through the inherent selectivity of the thermal reaction and the subsequent crystallization step. Since no external catalysts are introduced, there is no risk of metal contamination, which is a common issue in catalytic hydrogenation or coupling reactions. The major potential impurities would be unreacted starting materials or over-reacted species, both of which are effectively removed during the recrystallization process. The patent specifies the use of solvents like ethyl acetate, methanol, or acetonitrile for purification, where the solubility differences between the product and impurities are exploited. Heating the crude solid to dissolve it followed by controlled cooling allows the high-purity Compound II to crystallize out, leaving soluble impurities in the mother liquor. This physical separation mechanism ensures the final product meets the stringent >99% purity requirement essential for reference standards.
How to Synthesize Pregabalin Impurity Compound II Efficiently
The synthesis protocol outlined in the patent offers a straightforward pathway for laboratories and manufacturing units to produce this critical reference standard. The process begins with the precise weighing of phenylethylamine and urea, which are combined in a reaction vessel. The mixture is then subjected to controlled heating to initiate the condensation reaction. Following the reaction period, the mixture is cooled to induce solidification, and the crude product is isolated via filtration. To achieve the requisite analytical grade purity, the crude solid undergoes a recrystallization step using a selected organic solvent. For a detailed breakdown of the specific parameters, equipment requirements, and safety considerations, please refer to the standardized synthesis guide below.
- Mix phenylethylamine and urea in a molar ratio of 1: 2.0 to 1:3 without adding external solvents or catalysts.
- Heat the reaction mixture to 110-120°C and maintain for 3-5 hours to facilitate thermal condensation.
- Cool the mixture, filter the crude solid, and recrystallize using ethyl acetate or methanol to achieve >99% purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this solvent-free technology offers tangible benefits beyond mere technical feasibility. The elimination of solvents and catalysts translates directly into reduced raw material costs and lower waste disposal expenses. Traditional synthesis methods often incur significant hidden costs associated with solvent recovery, distillation, and the treatment of hazardous waste streams. By removing these unit operations, the overall cost of goods sold (COGS) for the impurity standard is significantly optimized. Furthermore, the simplified workflow reduces the dependency on complex supply chains for specialized catalysts or high-grade anhydrous solvents, thereby enhancing supply chain resilience and reducing lead times for production batches.
- Cost Reduction in Manufacturing: The process eliminates the need for expensive transition metal catalysts and large volumes of organic solvents during the reaction phase. This reduction in material input directly lowers the variable costs associated with production. Additionally, the simplified workup procedure requires less energy for solvent removal and recovery, contributing to substantial operational expenditure savings. The high yield reported in the patent examples further maximizes the output per batch, ensuring efficient utilization of raw materials and reducing the cost per gram of the final high-purity product.
- Enhanced Supply Chain Reliability: Relying on commodity chemicals like phenylethylamine and urea mitigates the risk of supply disruptions often associated with specialized reagents. The robustness of the solvent-free method means that production is less sensitive to variations in solvent quality or availability. This stability allows for more predictable production scheduling and inventory management. For pharmaceutical companies requiring consistent supplies of reference standards for long-term stability studies, this reliability is crucial for maintaining uninterrupted quality control operations and regulatory compliance.
- Scalability and Environmental Compliance: The absence of volatile solvents during the reaction significantly improves the safety profile of the manufacturing process, reducing the risk of fire and explosion hazards. This makes the process easier to scale from laboratory to pilot and commercial scales without requiring extensive modifications to safety infrastructure. Moreover, the reduced generation of hazardous waste aligns with increasingly strict environmental regulations, facilitating smoother permitting processes and enhancing the corporate sustainability profile of the manufacturer. The ability to produce high-purity materials with a minimal environmental footprint is a distinct competitive advantage in the modern green chemical market.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Pregabalin Impurity II. These answers are derived directly from the technical specifications and experimental data provided in patent CN113804805A. Understanding these details is essential for R&D teams integrating this standard into their quality control workflows and for procurement teams evaluating supplier capabilities.
Q: What is the primary advantage of this synthesis method for Impurity II?
A: The primary advantage is the elimination of organic solvents and catalysts during the reaction phase, utilizing the raw material phenylethylamine as the reaction medium itself. This significantly reduces waste generation and simplifies the downstream purification process while maintaining yields above 98%.
Q: Why is high-purity Impurity II critical for Pregabalin manufacturing?
A: Impurity II is a known byproduct formed during the synthesis of the key Pregabalin intermediate (Compound III). Regulatory guidelines require strict control of this impurity (typically <0.5%). Having a certified high-purity reference standard (>99%) is essential for accurate HPLC quantification and ensuring the safety of the final API.
Q: Can this process be scaled for industrial reference standard production?
A: Yes, the patent demonstrates scalability from gram scale (10g) to kilogram scale (2.0kg) with consistent results. The absence of complex catalytic systems and the use of simple recrystallization make it highly suitable for commercial scale-up of complex pharmaceutical reference materials.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Pregabalin Impurity II Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality reference standards play in the development and manufacturing of safe pharmaceutical products. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your demand for Pregabalin Impurity II with consistency and precision. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of our reference materials meets the highest international standards. Our commitment to green chemistry and process efficiency allows us to deliver superior products that support your regulatory filings and quality assurance programs.
We invite you to collaborate with us to optimize your supply chain for critical impurity standards. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our advanced solvent-free technology can enhance your quality control capabilities while reducing overall costs.