Advanced Solvent-Free Synthesis of Pregabalin Impurity II for Global Quality Control Standards
The pharmaceutical industry faces rigorous challenges in maintaining the purity and safety of Active Pharmaceutical Ingredients (APIs), particularly for widely prescribed medications like Pregabalin. Patent CN113804805B introduces a groundbreaking methodology for the preparation of Pregabalin intermediate impurity Compound II, addressing a critical gap in quality control infrastructure. As regulatory bodies demand stricter impurity profiling, the ability to synthesize high-purity reference standards efficiently becomes a cornerstone of compliant manufacturing. This patent details a novel, environmentally benign route that bypasses traditional solvent-heavy processes, offering a robust solution for generating the essential calibration standards needed to monitor trace contaminants in the final drug product.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
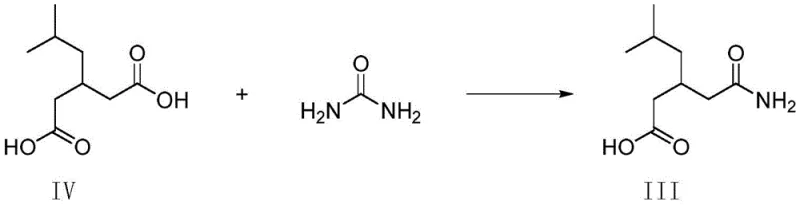
Traditionally, the presence of Compound II in Pregabalin synthesis has been an uncontrolled and problematic occurrence rather than a deliberate synthesis target. In standard manufacturing routes, Compound III is a key intermediate prepared by reacting Compound IV with urea. However, Compound IV is often derived from mother liquors containing residual R-phenylethylamine from previous resolution steps. This residual amine inadvertently reacts with urea to form Compound II, an impurity that persists through subsequent reactions. The derivative impurities formed from Compound II are notoriously difficult to remove via conventional extraction, washing, or crystallization techniques, often requiring complex and costly purification protocols to meet safety specifications. Furthermore, isolating pure Compound II from these complex reaction mixtures for use as a reference standard is inefficient and yields inconsistent results.
The Novel Approach
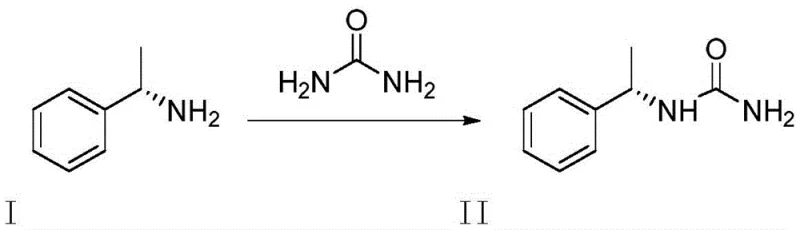
In stark contrast to the accidental and messy formation in traditional routes, the method disclosed in CN113804805B offers a direct, intentional, and highly efficient synthesis of Compound II. By reacting phenylethylamine directly with urea under solvent-free conditions, the process achieves exceptional conversion rates without the need for external catalysts or diluents. The reaction utilizes the reactant itself as the solvent, creating a high-concentration environment that drives the equilibrium towards the desired urea derivative. Following the thermal reaction, a simple recrystallization step using common organic solvents like ethyl acetate yields the target impurity with purity levels exceeding 99%. This deliberate synthesis strategy transforms a problematic contaminant into a manageable, high-value reference material, streamlining the quality assurance workflow for Pregabalin manufacturers globally.
Mechanistic Insights into Solvent-Free Urea Condensation
The core chemical transformation relies on a nucleophilic attack mechanism where the primary amine group of phenylethylamine attacks the electrophilic carbonyl carbon of the urea molecule. Under elevated temperatures ranging from 90°C to 150°C, the kinetic energy of the molecules overcomes the activation barrier, facilitating the displacement of ammonia and the formation of the stable N-substituted urea bond. The absence of solvent is not merely a cost-saving measure but a strategic mechanistic advantage; it maximizes the collision frequency between reactant molecules, significantly accelerating the reaction rate compared to diluted solutions. This high-concentration regime ensures that the reaction proceeds to near-completion within a relatively short timeframe of 3 to 5 hours, minimizing the formation of side products that typically arise in prolonged or dilute reactions.
Impurity control in this synthesis is inherently managed by the stoichiometry and the physical properties of the product. By optimizing the molar ratio of phenylethylamine to urea, specifically within the range of 1:2.0 to 1:3.0, the process ensures that urea is in excess, driving the consumption of the amine and preventing the accumulation of unreacted starting material which could complicate purification. The resulting Compound II exhibits distinct solubility characteristics that allow for effective separation from any remaining urea or minor byproducts through temperature-controlled crystallization. This precise control over the reaction environment and downstream processing guarantees a consistent impurity profile, which is vital when the product itself is intended to serve as a standard for detecting trace levels of the same compound in other batches.
How to Synthesize N-(1-phenylethyl)urea Efficiently
The synthesis protocol outlined in the patent provides a straightforward pathway for laboratories and production facilities to generate high-purity Compound II. The process begins with the direct mixing of the amine and urea, eliminating the need for complex solvent handling systems. Detailed operational parameters regarding temperature ramps and crystallization kinetics are critical for achieving the reported yields of approximately 98%. For teams looking to implement this reference standard production, the following guide summarizes the critical operational phases derived from the patent examples.
- Mix phenylethylamine and urea in a reaction vessel without additional solvents, utilizing the amine as the reaction medium.
- Heat the mixture to 110°C–120°C and maintain for 3–5 hours to drive the condensation reaction to completion.
- Cool the reaction, filter the crude solid, and recrystallize using ethyl acetate or methanol to achieve >99% purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this solvent-free technology represents a significant opportunity to optimize operational expenditures and reduce logistical complexity. The elimination of bulk organic solvents from the reaction phase drastically cuts down on raw material procurement costs and removes the associated hazards of storing and handling large volumes of flammable liquids. Furthermore, the simplified workflow reduces the burden on waste management systems, as there is significantly less solvent waste to treat or dispose of, aligning production practices with increasingly stringent environmental regulations. This lean manufacturing approach translates directly into lower unit costs for producing essential reference standards, allowing pharmaceutical companies to allocate resources more effectively across their R&D portfolios.
- Cost Reduction in Manufacturing: The removal of catalysts and reaction solvents creates a fundamentally cheaper production model. By relying on thermal energy rather than expensive reagents to drive the reaction, the variable costs per kilogram of product are substantially reduced. Additionally, the high yield reported in the patent examples means that less raw material is wasted, maximizing the return on investment for every batch produced. This efficiency is crucial for maintaining competitive pricing in the global market for pharmaceutical intermediates and reference materials.
- Enhanced Supply Chain Reliability: The raw materials required for this synthesis, phenylethylamine and urea, are commodity chemicals with robust and stable global supply chains. Unlike specialized catalysts or exotic solvents that may face availability fluctuations, these inputs are readily accessible from multiple suppliers worldwide. This abundance ensures that production schedules remain uninterrupted, mitigating the risk of delays in generating critical quality control standards. Reliable access to these standards is essential for maintaining continuous API manufacturing without regulatory bottlenecks.
- Scalability and Environmental Compliance: The simplicity of the reaction setup—essentially a heated vessel followed by filtration—makes this process exceptionally easy to scale from laboratory grams to industrial tons. There are no complex exothermic risks associated with rapid catalyst addition or sensitive moisture-sensitive reagents. Moreover, the green chemistry credentials of a solvent-free process enhance the corporate sustainability profile, helping organizations meet their carbon reduction goals and comply with green manufacturing mandates without sacrificing product quality or throughput.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis method. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the feasibility and benefits of adopting this technology for quality control applications.
Q: Why is controlling Compound II critical in Pregabalin manufacturing?
A: Compound II forms as a byproduct when residual R-phenylethylamine reacts with urea. It derivatives into difficult-to-remove impurities that compromise final API safety, necessitating strict limits (≤0.5%) and high-purity reference standards for HPLC quantification.
Q: What are the advantages of the solvent-free method described in CN113804805B?
A: The method eliminates the need for external organic solvents and catalysts, significantly reducing waste generation and raw material costs while simplifying the downstream purification process through direct crystallization.
Q: Can this synthesis method be scaled for industrial reference standard production?
A: Yes, the process utilizes simple heating and filtration steps with high yields (approx. 98%), making it highly scalable and suitable for producing large batches of certified reference materials required for regulatory compliance.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Pregabalin Impurity II Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your final pharmaceutical product depends on the precision of your analytical controls. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your demand for high-purity reference standards with consistency and speed. We utilize stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Pregabalin Impurity II we supply meets the exacting requirements of international pharmacopoeias, enabling you to validate your manufacturing processes with confidence.
We invite you to collaborate with us to secure a stable supply of this critical intermediate impurity standard. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our advanced synthesis capabilities can support your quality assurance objectives and streamline your supply chain operations.