Scalable Asymmetric Synthesis of Benzoxazinone HIV Inhibitor Intermediates for Commercial Production
Scalable Asymmetric Synthesis of Benzoxazinone HIV Inhibitor Intermediates for Commercial Production
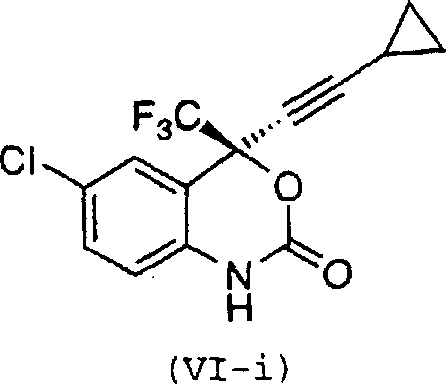
The pharmaceutical industry continuously seeks robust and scalable pathways for the production of complex active pharmaceutical ingredients (APIs) and their critical intermediates. Patent CN1251582A discloses a groundbreaking asymmetric synthesis method for (S)-6-chloro-4-cyclopropylethynyl-4-trifluoromethyl-1,4-dihydro-2H-3,1-benzoxazin-2-one, a potent non-nucleoside human immunodeficiency virus (HIV) reverse transcriptase inhibitor. This compound represents a vital class of therapeutics capable of interrupting viral replication, yet its historical production has been plagued by inefficiencies and environmental hazards. The disclosed innovation introduces a novel sequence of reactions that not only enhances stereochemical control but also fundamentally restructures the manufacturing workflow to favor industrial viability. By replacing hazardous oxidants and eliminating chromatographic purification steps, this technology offers a compelling value proposition for reliable pharmaceutical intermediate supplier networks aiming to optimize their supply chains. The following analysis details the technical merits and commercial implications of this advanced synthetic route.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of benzoxazinone derivatives relied heavily on methodologies described in earlier literature, such as those by Thompson et al. and European Patent Application 582,455 A1. These conventional routes typically involved the metallation of p-chloroaniline pivalamide followed by nucleophilic substitution and Grignard addition. A significant bottleneck in these legacy processes was the debenzylation step, which necessitated the use of ceric ammonium nitrate. This reagent is not only highly toxic but also generates substantial quantities of waste liquid containing heavy metal cerium ions, creating severe environmental compliance challenges and escalating waste disposal costs. Furthermore, these older methods often required inefficient chromatographic purification steps to isolate optical isomers, which drastically reduces overall throughput and increases the cost of goods sold (COGS). The reliance on unstable reagents and the generation of hazardous byproducts rendered these processes suboptimal for modern, green chemistry-focused manufacturing environments.
The Novel Approach
In stark contrast, the novel approach detailed in the patent data introduces a streamlined five-step synthesis that addresses these critical pain points through innovative chemical engineering. The process initiates with an acid-catalyzed benzylation using benzyl alcohols instead of the more expensive and unstable benzyl chloride analogues, facilitating a smoother reaction profile. Crucially, the route employs a chiral induction strategy using (1R,2S)-pyrrolidinyl norephedrine to achieve high enantiomeric excess during the acetylide addition, effectively bypassing the need for resolution via chromatography. The subsequent oxidative cyclization and debenzylation steps utilize safer reagents like DDQ or p-chloranil and sodium borohydride trapping, completely eliminating the heavy metal waste associated with ceric ammonium nitrate. This paradigm shift allows for the isolation of stable solid intermediates via recrystallization, significantly enhancing process robustness. 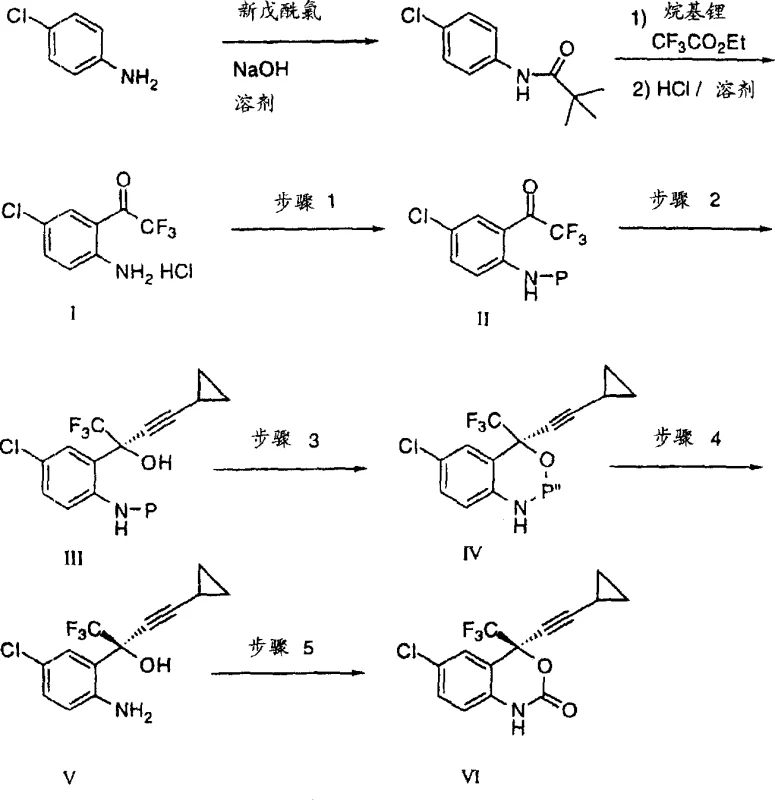
Mechanistic Insights into Chiral Induction and Oxidative Cyclization
The core of this synthetic breakthrough lies in the precise control of stereochemistry during the formation of the quaternary carbon center. The mechanism involves the in situ generation of lithium cyclopropylacetylide, which is then reacted with the protected ketone intermediate in the presence of the chiral inducer (1R,2S)-pyrrolidinyl norephedrine. This chiral ligand coordinates with the lithium species, creating a rigid transition state that directs the nucleophilic attack of the acetylide anion to the prochiral carbonyl carbon with high facial selectivity. This step is critical for establishing the (S)-configuration required for biological activity. Following the formation of the tertiary alcohol, the process moves to oxidative cyclization. Here, the protected amine is oxidized to an imine species using reagents such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ). This oxidation triggers an intramolecular nucleophilic attack by the hydroxyl group onto the imine carbon, closing the benzoxazine ring. The mechanistic elegance of this sequence ensures that the stereochemical integrity established in the previous step is maintained throughout the ring closure, yielding the desired diastereomer with high fidelity.
Impurity control is another cornerstone of this mechanism, particularly during the debenzylation phase. When the benzyl protecting group is cleaved under basic conditions, it generates an aromatic aldehyde byproduct (e.g., p-methoxybenzaldehyde) which is highly reactive and could potentially condense with the free amine of the product, forming difficult-to-remove Schiff base impurities. The novel method ingeniously mitigates this risk by introducing a trapping agent, specifically sodium borohydride, into the reaction mixture. This reducing agent selectively converts the liberated aldehyde into its corresponding alcohol immediately upon formation. By chemically neutralizing the reactive byproduct in situ, the process prevents side reactions that would otherwise compromise the purity of the final amino alcohol intermediate. This strategic use of a scavenger ensures that the crude product obtained is of sufficient quality to proceed directly to the final cyclization step, often requiring only simple crystallization rather than complex purification workflows.
How to Synthesize (S)-6-chloro-4-cyclopropylethynyl-4-trifluoromethyl-1,4-dihydro-2H-3,1-benzoxazin-2-one Efficiently
The execution of this synthesis requires careful attention to reaction conditions, particularly temperature control and reagent stoichiometry, to maximize yield and enantiomeric purity. The process is designed to be telescoped where possible, minimizing the handling of sensitive intermediates. For instance, the preparation of the lithium acetylide reagent is best performed as a solution stream to ensure consistent concentration and reactivity before it is introduced to the ketone substrate. Similarly, the oxidative cyclization can be optimized by selecting solvents like heptane or ethyl acetate which facilitate the precipitation of byproducts, simplifying the workup. The detailed standardized synthesis steps below outline the critical parameters for each stage of the transformation, providing a roadmap for technical teams to replicate this high-efficiency route in a pilot or production setting.
- Perform acid-catalyzed benzylation of the substituted aniline with p-methoxybenzyl alcohol to form the protected amine intermediate.
- Execute chiral induction by reacting the protected ketone with lithium cyclopropylacetylide in the presence of (1R,2S)-pyrrolidinyl norephedrine.
- Conduct oxidative cyclization using an oxidant like DDQ or p-chloranil to form the benzoxazine ring structure.
- Remove the protecting group via base-mediated cleavage with simultaneous trapping of the aldehyde byproduct using sodium borohydride.
- Complete the synthesis by cyclizing the amino alcohol intermediate with phosgene to yield the final benzoxazinone product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route translates into tangible operational improvements and risk mitigation. The elimination of chromatographic purification is perhaps the most significant economic driver, as column chromatography is notoriously difficult to scale and represents a major bottleneck in batch processing times. By relying on crystallization for purification, the process becomes inherently more scalable and compatible with standard stainless steel reactor infrastructure found in most multipurpose chemical plants. Furthermore, the removal of ceric ammonium nitrate from the process map drastically reduces the regulatory burden associated with heavy metal waste disposal. This not only lowers direct disposal costs but also minimizes the risk of supply chain disruptions caused by stringent environmental audits or changes in waste treatment regulations. The ability to produce stable solid intermediates also enhances inventory management, allowing for the strategic stocking of key precursors to buffer against raw material volatility.
- Cost Reduction in Manufacturing: The streamlined workflow significantly lowers the cost of goods sold by removing expensive and time-consuming purification steps. The replacement of hazardous oxidants with safer alternatives reduces the need for specialized containment equipment and lowers insurance premiums related to chemical handling. Additionally, the high yield and selectivity of the chiral induction step minimize the loss of valuable starting materials, ensuring that raw material costs are optimized. The overall efficiency gains mean that less solvent and energy are consumed per kilogram of product, contributing to a leaner and more cost-effective manufacturing operation that improves margin potential for the final API.
- Enhanced Supply Chain Reliability: The robustness of this synthesis enhances supply continuity by reducing the number of failure points in the production line. The use of stable intermediates that can be isolated and stored provides flexibility in scheduling, decoupling upstream and downstream processes. Moreover, the reagents employed, such as p-methoxybenzyl alcohol and common alkali bases, are commodity chemicals with reliable global availability, reducing the risk of sourcing bottlenecks. This stability ensures that production timelines can be met consistently, which is critical for maintaining the supply of life-saving HIV medications to patients worldwide without interruption due to technical manufacturing issues.
- Scalability and Environmental Compliance: This process is explicitly designed for commercial scale-up, moving seamlessly from kilogram to multi-ton production without the need for process re-engineering. The avoidance of heavy metals aligns perfectly with modern green chemistry initiatives and corporate sustainability goals, making the supply chain more resilient to future regulatory tightening. The simplified waste profile facilitates easier permitting and operation in diverse geographic regions, allowing for a more distributed and secure manufacturing network. This environmental stewardship not only protects the ecosystem but also safeguards the brand reputation of the pharmaceutical company by ensuring ethical and compliant production practices.
Frequently Asked Questions (FAQ)
The following questions address common technical and operational inquiries regarding the implementation of this asymmetric synthesis technology. These insights are derived directly from the patent specifications and are intended to clarify the feasibility and advantages of the method for technical decision-makers. Understanding these nuances is essential for evaluating the integration of this route into existing manufacturing portfolios.
Q: How does this novel synthesis route improve environmental compliance compared to conventional methods?
A: The novel method eliminates the use of highly toxic ceric ammonium nitrate used in traditional debenzylation steps, thereby removing heavy metal cerium ions from the waste stream and significantly reducing hazardous waste disposal costs.
Q: What strategies are employed to ensure high enantiomeric purity without chromatography?
A: The process utilizes a highly selective chiral inducer, (1R,2S)-pyrrolidinyl norephedrine, during the acetylide addition step, combined with efficient recrystallization protocols for intermediates, allowing for the isolation of enantiomerically pure products without the need for expensive chromatographic separation.
Q: Is this synthetic pathway suitable for large-scale industrial manufacturing?
A: Yes, the methodology is specifically designed for scalability, featuring stable solid intermediates that can be purified by recrystallization and solution-stream processing for sensitive reagents, making it robust for kilogram to multi-ton production scales.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Benzoxazinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and compliant synthesis routes for complex pharmaceutical intermediates like benzoxazinones. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial reality is seamless. We are committed to delivering high-purity intermediates that meet stringent purity specifications, utilizing rigorous QC labs to verify every batch. Our facility is equipped to handle the specific requirements of this novel synthesis, including the safe handling of organolithium reagents and the implementation of advanced crystallization techniques to guarantee product quality. By leveraging our expertise, partners can accelerate their development timelines and secure a stable supply of critical HIV inhibitor intermediates.
We invite you to engage with our technical procurement team to discuss how this advanced synthetic route can be tailored to your specific production needs. Request a Customized Cost-Saving Analysis today to understand the potential economic impact of switching to this greener, more efficient methodology. Our experts are ready to provide specific COA data and route feasibility assessments to support your supply chain optimization strategies. Let us help you build a more resilient and cost-effective supply chain for your next-generation antiviral therapies.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →