Advanced Manufacturing of Prostaglandin F Derivatives for Glaucoma Treatment via Selective Silylation
Advanced Manufacturing of Prostaglandin F Derivatives for Glaucoma Treatment via Selective Silylation
The pharmaceutical landscape for treating glaucoma and intraocular hypertension relies heavily on the consistent supply of high-purity prostaglandin derivatives. Patent CN1990450A introduces a sophisticated synthetic methodology for preparing prostaglandin F type derivatives, specifically targeting the structural motif shown in Formula (I). This patent represents a significant advancement in process chemistry by addressing the longstanding challenges of selectivity and purification in complex lipid-like molecules. The core innovation lies in a novel intermediate strategy that employs a dual-silylation protection scheme, distinguishing between bulky triethylsilyl groups for stability and smaller trimethylsilyl groups for purification efficiency. 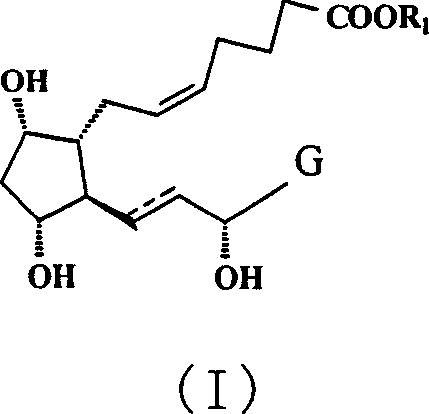 For procurement managers and R&D directors, understanding this pathway is critical, as it offers a robust route to scalable production of these high-value ophthalmic intermediates, ensuring both cost-effectiveness and stringent quality control standards required for clinical applications.
For procurement managers and R&D directors, understanding this pathway is critical, as it offers a robust route to scalable production of these high-value ophthalmic intermediates, ensuring both cost-effectiveness and stringent quality control standards required for clinical applications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for prostaglandin analogs often suffer from poor chemoselectivity during the functionalization of the cyclopentane ring and the aliphatic side chains. Conventional protection strategies frequently utilize a single type of protecting group throughout the synthesis, which can lead to significant difficulties during the purification of intermediates, particularly after carbon-carbon bond-forming reactions like the Wittig olefination. The presence of highly polar byproducts, such as phosphine oxides and unreacted phosphonium salts, often necessitates cumbersome chromatographic separations that are difficult to scale and result in substantial yield losses. Furthermore, standard reduction conditions for lactone intermediates can inadvertently affect unprotected hydroxyl groups or cause epimerization at chiral centers if the protection is not sufficiently robust, leading to complex impurity profiles that are costly to remove and detrimental to the final API safety profile.
The Novel Approach
The methodology disclosed in CN1990450A overcomes these hurdles through a strategic, step-wise protection protocol that optimizes both reaction stability and downstream workup efficiency. As illustrated in the overall reaction scheme, the process begins with the installation of bulky triethylsilyl (TES) groups, which provide steric shielding during the subsequent reduction and olefination steps. 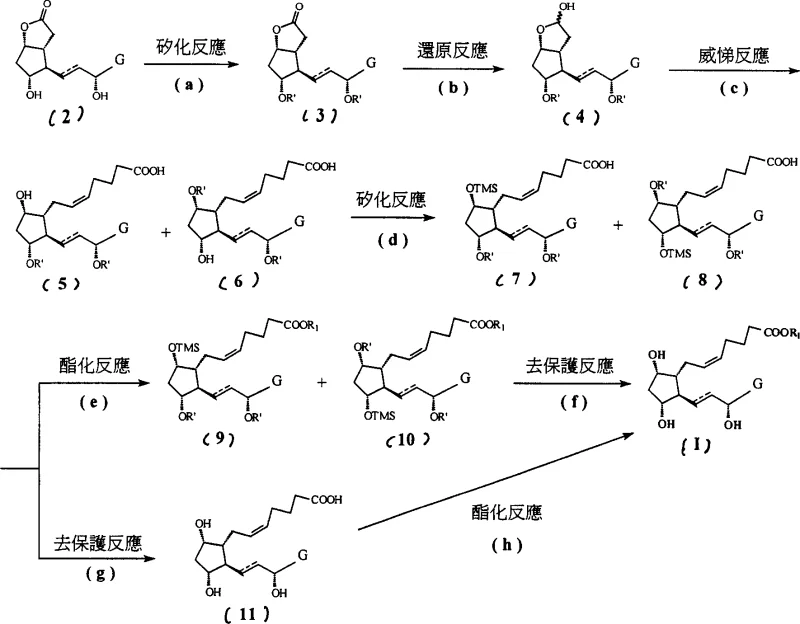 Crucially, the process introduces a second silylation step using trimethylsilyl chloride (TMSCl) after the Wittig reaction. This specific modification increases the lipophilicity of the intermediate just enough to allow for the effective extraction of polar impurities into the aqueous phase, a purification technique that is far more scalable and cost-effective than column chromatography. This novel approach transforms a potentially messy synthesis into a streamlined, industrially viable process that significantly enhances the overall yield and purity of the final prostaglandin F derivative.
Crucially, the process introduces a second silylation step using trimethylsilyl chloride (TMSCl) after the Wittig reaction. This specific modification increases the lipophilicity of the intermediate just enough to allow for the effective extraction of polar impurities into the aqueous phase, a purification technique that is far more scalable and cost-effective than column chromatography. This novel approach transforms a potentially messy synthesis into a streamlined, industrially viable process that significantly enhances the overall yield and purity of the final prostaglandin F derivative.
Mechanistic Insights into Selective Silylation and Wittig Olefination
The chemical elegance of this process is rooted in the orthogonal use of silyl protecting groups to manage reactivity and solubility. In the initial stage, the reaction of the starting lactone with triethylsilyl chloride (TES-Cl) in the presence of a base like triethylamine creates a sterically hindered environment around the cyclopentane hydroxyls. This bulk is essential during the subsequent reduction with diisobutylaluminium hydride (DIBAL-H), as it prevents the reducing agent from interacting with the protected oxygen atoms, thereby directing the reduction exclusively to the lactone carbonyl to form the lactol intermediate. The stability of the TES group under these cryogenic conditions (-60 to -70°C) ensures that the delicate stereochemistry of the ring system is preserved, preventing the formation of diastereomeric impurities that could compromise the biological activity of the final drug substance.
Following the Wittig reaction, which extends the upper side chain, the process employs a second silylation using the smaller trimethylsilyl (TMS) group. 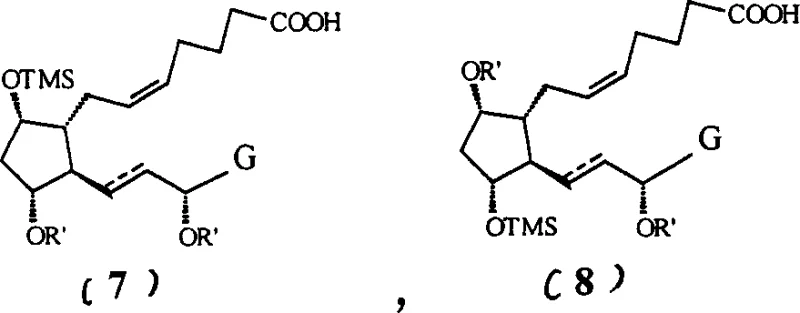 This step is mechanistically distinct from the first; while the TES group was chosen for stability, the TMS group is selected for its ability to modulate physicochemical properties without adding excessive steric bulk. By converting the newly exposed hydroxyls to TMS ethers, the intermediate becomes sufficiently lipophilic to partition into organic solvents during aqueous workups, leaving behind the highly polar phosphonium salt byproducts generated during the Wittig coupling. This solubility switch is a critical process parameter that simplifies isolation, reduces solvent consumption, and minimizes the risk of carryover impurities into the final esterification and deprotection stages, ultimately delivering a product with a superior impurity profile.
This step is mechanistically distinct from the first; while the TES group was chosen for stability, the TMS group is selected for its ability to modulate physicochemical properties without adding excessive steric bulk. By converting the newly exposed hydroxyls to TMS ethers, the intermediate becomes sufficiently lipophilic to partition into organic solvents during aqueous workups, leaving behind the highly polar phosphonium salt byproducts generated during the Wittig coupling. This solubility switch is a critical process parameter that simplifies isolation, reduces solvent consumption, and minimizes the risk of carryover impurities into the final esterification and deprotection stages, ultimately delivering a product with a superior impurity profile.
How to Synthesize Prostaglandin F Derivatives Efficiently
The synthesis of these complex molecules requires precise control over reaction parameters to ensure reproducibility and high quality. The patented process outlines a clear sequence of protection, reduction, chain extension, and final functionalization that can be adapted for commercial scale-up. Operators must pay close attention to the stoichiometry of the silylating agents and the temperature gradients during the reduction phase to maximize conversion. The following guide summarizes the critical operational phases derived from the patent examples, providing a framework for implementing this technology in a GMP environment. Detailed standardized synthesis steps are provided in the section below.
- Perform initial protection of the cyclopentane ring hydroxyls using triethylsilyl chloride (TES-Cl) to prevent side reactions during reduction.
- Reduce the lactone carbonyl group to a lactol using DIBAL-H at low temperatures (-60 to -70°C) to maintain stereochemical integrity.
- Execute a Wittig reaction with a phosphonium ylide to extend the upper side chain, followed by a second silylation with TMSCl to facilitate purification.
- Conclude with esterification of the carboxylic acid tail and global deprotection to yield the final Prostaglandin F type derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this synthetic route offers tangible benefits beyond mere chemical yield. The strategic design of the process directly addresses key cost drivers in fine chemical manufacturing, specifically regarding raw material utilization and waste management. By enabling purification through liquid-liquid extraction rather than relying heavily on preparative chromatography, the process drastically reduces the consumption of silica gel and organic solvents, which are significant cost centers in large-scale production. This efficiency translates into a more competitive cost structure for the final intermediate, allowing downstream API manufacturers to optimize their margins while maintaining high quality standards.
- Cost Reduction in Manufacturing: The elimination of extensive chromatographic purification steps results in substantial cost savings by reducing solvent usage and processing time. The ability to remove polar impurities via simple extraction after the second silylation step streamlines the workflow, lowering the operational expenditure associated with waste disposal and solvent recovery. Furthermore, the use of commercially available and inexpensive silylating reagents like TES-Cl and TMSCl ensures that raw material costs remain stable and predictable, avoiding the volatility associated with specialized or proprietary catalysts.
- Enhanced Supply Chain Reliability: The reliance on standard chemical reagents and well-established reaction types, such as the Wittig reaction and silyl ether protection, mitigates the risk of supply disruptions. Unlike processes dependent on scarce transition metal catalysts or bespoke enzymes, this route utilizes commodity chemicals that are readily available from multiple global suppliers. This diversification of the supply base enhances resilience against market fluctuations and ensures continuous production capability, which is vital for meeting the demanding delivery schedules of the pharmaceutical industry.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, with reaction conditions that are manageable in large reactors, such as the controlled low-temperature reduction and ambient temperature silylations. The reduction in solvent intensity and the avoidance of heavy metal catalysts align with green chemistry principles, simplifying environmental compliance and reducing the regulatory burden associated with residual metal testing in the final product. This environmental compatibility facilitates smoother regulatory filings and supports the sustainability goals of modern pharmaceutical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this prostaglandin synthesis technology. These answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for potential partners. Understanding these details is essential for evaluating the feasibility of integrating this process into existing manufacturing lines.
Q: Why is a dual silylation strategy employed in this synthesis?
A: The process utilizes triethylsilyl (TES) groups initially for their stability during the harsh reduction conditions, followed by trimethylsilyl (TMS) groups later to increase lipophilicity, allowing for the efficient removal of polar phosphonium salt impurities via extraction.
Q: What are the critical temperature controls required for the reduction step?
A: The reduction of the lactone to the lactol using DIBAL-H requires strict temperature control between -60°C and -70°C to prevent over-reduction and ensure the formation of the correct hemiacetal intermediate without degrading the sensitive cyclopentane core.
Q: How does this process improve supply chain reliability for ophthalmic APIs?
A: By utilizing common silicification reagents like TES-Cl and TMSCl and avoiding exotic catalysts, the process ensures raw material availability. Furthermore, the enhanced purification capability via lipophilicity adjustment reduces batch failure rates, ensuring consistent supply continuity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Prostaglandin F Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust synthetic pathways in the production of life-saving ophthalmic medications. Our team of expert chemists has extensively analyzed the methodology described in CN1990450A and possesses the technical capability to execute this complex multi-step synthesis with precision. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and reliability. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of prostaglandin intermediate meets the highest international standards for safety and efficacy.
We invite you to collaborate with us to leverage this advanced technology for your next project. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact us today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our manufacturing expertise can drive value and efficiency in your supply chain for high-purity prostaglandin derivatives.