Advanced Organocatalytic Synthesis of Bicyclic Proline Esters for High-Purity API Manufacturing
The pharmaceutical industry constantly seeks robust and scalable pathways for critical active pharmaceutical ingredient (API) intermediates, particularly for antiviral treatments like Telaprevir. Patent CN103562182A discloses a groundbreaking seven-step synthetic methodology for producing specific ester compounds, designated generally as formula (1), which serve as pivotal building blocks in medicinal chemistry. This novel approach diverges significantly from traditional routes by leveraging asymmetric organocatalysis to construct complex bicyclic frameworks with high stereochemical fidelity. The process begins with simple aldehyde precursors and systematically builds molecular complexity through nitroaldol reactions, protective group manipulations, and cyclization events. By integrating water and carboxylic acid additives in the initial catalytic step, the invention achieves remarkable reaction rates and yields that were previously unattainable with stoichiometric chiral auxiliaries. This technical advancement represents a significant leap forward for manufacturers aiming to secure a reliable pharmaceutical intermediates supplier capable of delivering high-purity materials consistently.
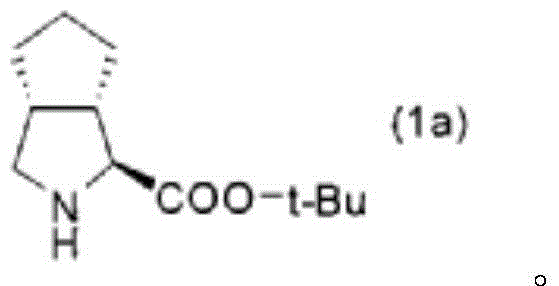
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods, such as those described in WO2007/022459 and WO2010/126881, typically rely on the derivatization of 3-azabicyclo[3.3.0]nonane hydrochloride. While effective, these conventional pathways often suffer from significant drawbacks regarding raw material availability and cost efficiency. Starting with pre-formed bicyclic amine salts limits flexibility and can introduce supply chain bottlenecks if the specific salt is not commercially abundant. Furthermore, achieving the necessary stereochemical purity often requires extensive chromatographic purification or resolution steps, which drastically reduce overall process mass intensity and increase waste generation. The reliance on stoichiometric chiral reagents in older methodologies also inflates the cost of goods sold (COGS), making the final API less competitive in price-sensitive markets. Additionally, harsh reaction conditions sometimes required in these legacy processes can pose safety risks and complicate the engineering controls needed for large-scale manufacturing.
The Novel Approach
In stark contrast, the methodology outlined in CN103562182A initiates synthesis from accessible aldehyde compounds, such as 1-formylcyclopentene, reacting them with nitromethane under organocatalytic conditions. This strategy fundamentally shifts the paradigm from modifying complex cores to constructing them with precision. The use of a chiral pyrrolidine catalyst, specifically compounds of formula (9), enables the direct installation of chirality at the beta-position of the nitroaldehyde intermediate with exceptional enantioselectivity. This eliminates the need for downstream resolution, thereby streamlining the entire production workflow. The process is designed with scalability in mind, utilizing common solvents like alcohols and ethers, and operating within mild temperature ranges typically between -20°C and 50°C. By avoiding expensive transition metals and stoichiometric chiral sources, this novel approach offers a pathway for substantial cost reduction in pharmaceutical intermediates manufacturing while maintaining the rigorous purity standards required for clinical applications.
Mechanistic Insights into Organocatalytic Nitroaldol Reaction
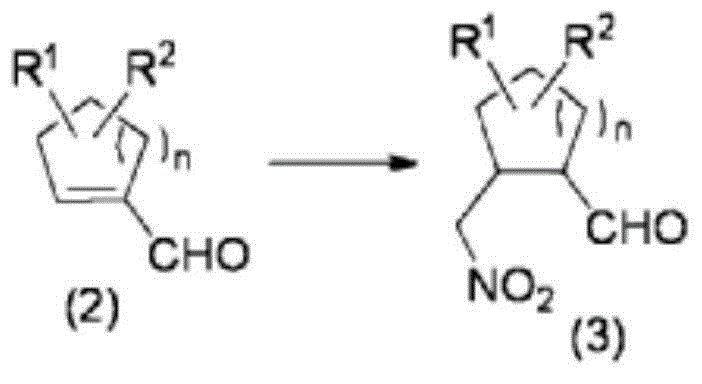
The cornerstone of this synthetic route is Step A, where the aldehyde compound (2) undergoes a Henry reaction (nitroaldol reaction) with nitromethane. This transformation is mediated by a chiral pyrrolidine catalyst, which activates the aldehyde through iminium ion formation or enamine catalysis, depending on the specific mechanistic cycle. The presence of the bulky diphenyltrimethylsilyloxymethyl group on the catalyst creates a defined chiral environment that directs the attack of the nitromethane anion to a specific face of the aldehyde. Crucially, the patent highlights the synergistic effect of adding water and carboxylic acids, such as acetic acid or benzoic acid, to the reaction mixture. These additives facilitate proton transfer steps and stabilize transition states, significantly accelerating the reaction rate even when catalyst loading is reduced to as low as 0.1 mol%. This mechanistic optimization ensures that the nitroaldehyde compound (3) is produced with high optical purity, setting the stereochemical trajectory for all subsequent steps in the synthesis.
Impurity control is meticulously managed throughout the sequence, particularly during the cyclization and deprotection phases. In Step E, the conversion of the protected aminoacetal to the nitrile involves an acid-mediated deprotection followed by base-induced cyclization. The choice of base, such as DBU or metal alkoxides, is critical to inducing the necessary stereochemical inversion at the alpha-carbon before cyanation. This precise control prevents the formation of diastereomeric impurities that could be difficult to separate later. Furthermore, the hydrolysis of the nitrile group in Step F is conducted under acidic conditions that are carefully tuned to convert the cyano group to a carboxylic acid without compromising the integrity of the amino protecting group, unless simultaneous deprotection is desired. This level of chemoselectivity minimizes the generation of side products, ensuring that the final ester compound (1) meets the stringent impurity profiles demanded by regulatory agencies for drug substance manufacturing.
How to Synthesize Bicyclic Proline Esters Efficiently
The synthesis of these high-value intermediates follows a logical progression of functional group transformations designed to maximize yield and stereochemical integrity. The process begins with the organocatalytic coupling of an aldehyde and nitromethane, followed by protection of the resulting aldehyde as an acetal to prevent side reactions during reduction. The nitro group is then reduced to a primary amine, which is subsequently protected to allow for selective manipulation of the acetal moiety. The core bicyclic structure is formed through a cascade involving acetal hydrolysis, base-mediated cyclization, and cyanation. Finally, the nitrile is hydrolyzed to the acid, and the amino protecting group is removed while simultaneously installing the final ester functionality. For detailed operational parameters, solvent choices, and workup procedures, please refer to the standardized synthesis guide below which encapsulates the critical process parameters identified in the patent examples.
- Step A: React aldehyde compound (2) with nitromethane using a pyrrolidine catalyst to form nitroaldehyde (3).
- Step B & C: Protect the aldehyde as an acetal (4) and reduce the nitro group to an amine (5).
- Step D & E: Protect the amine (6), deprotect acetal, and cyclize with a cyanating agent to form nitrile (7).
- Step F & G: Hydrolyze the nitrile to carboxylic acid (8) and perform deprotection/esterification to yield final ester (1).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers compelling strategic advantages beyond mere technical feasibility. The shift towards organocatalysis removes the dependency on precious metal catalysts, which are subject to volatile market pricing and strict residual metal limits in pharmaceutical products. By utilizing organic molecules as catalysts, the process inherently simplifies the purification train, reducing the need for expensive scavenging resins or complex extraction protocols. This simplification translates directly into lower operational expenditures and a reduced environmental footprint, aligning with modern green chemistry initiatives. Furthermore, the starting materials, including various cyclopentenecarbaldehydes and nitromethane, are commodity chemicals with robust global supply chains, mitigating the risk of raw material shortages that often plague specialized synthetic routes.
- Cost Reduction in Manufacturing: The implementation of this process drives down manufacturing costs primarily through the elimination of stoichiometric chiral reagents and the reduction of purification steps. Since the chirality is induced catalytically in the first step with high selectivity, there is no need for costly chiral chromatography or recrystallization resolutions later in the sequence. The ability to use water as a co-solvent in the initial step further reduces solvent consumption and waste disposal costs. Additionally, the mild reaction conditions minimize energy consumption associated with heating or cooling large reactors, contributing to a more economical production profile that enhances margin potential for the final API.
- Enhanced Supply Chain Reliability: Sourcing reliability is significantly improved because the synthesis relies on widely available bulk chemicals rather than custom-synthesized complex intermediates. The modular nature of the protecting group strategy allows manufacturers to adapt to fluctuations in the availability of specific reagents, such as switching between benzyl and tert-butyl protecting groups based on market conditions. The robustness of the reaction conditions, which tolerate the presence of water and operate at near-ambient temperatures, ensures consistent batch-to-batch quality even when scaling up from pilot plants to commercial facilities. This stability reduces the risk of batch failures and ensures a continuous flow of materials to downstream formulation teams.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, this route is exceptionally well-suited for large-scale production. The avoidance of heavy metals simplifies wastewater treatment and reduces the regulatory burden associated with metal residue testing. The use of common organic solvents like methanol, ethanol, and ethyl acetate facilitates solvent recovery and recycling, further enhancing the sustainability profile of the manufacturing process. The high yields reported in the patent examples across multiple steps indicate a convergent and efficient synthesis that minimizes waste generation per kilogram of product. This efficiency is crucial for meeting the increasing global demand for antiviral medications while adhering to increasingly strict environmental regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthetic technology. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on process capabilities and limitations. Understanding these nuances is essential for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the value proposition of intermediates produced via this method.
Q: What is the primary advantage of the organocatalytic Step A in this process?
A: The use of a chiral pyrrolidine catalyst in Step A allows for high stereoselectivity in the formation of the nitroaldehyde intermediate, eliminating the need for difficult chiral separations later in the synthesis.
Q: How does this method improve supply chain reliability compared to prior art?
A: By utilizing readily available starting materials like cyclopentenecarbaldehyde and nitromethane, rather than complex bicyclic amines, the process reduces dependency on scarce precursors and simplifies raw material sourcing.
Q: Can this process be scaled for commercial API production?
A: Yes, the reaction conditions are mild (typically 0°C to 30°C) and utilize standard organic solvents, making the transition from laboratory scale to multi-ton commercial production feasible without requiring specialized high-pressure equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Telaprevir Intermediate Supplier
The technological potential of this organocatalytic route is immense, offering a clear path to high-quality, cost-effective pharmaceutical intermediates. At NINGBO INNO PHARMCHEM, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. Our facility is equipped with rigorous QC labs and adheres to stringent purity specifications, guaranteeing that every batch of bicyclic proline ester meets the exacting standards required for API synthesis. We understand the critical nature of supply continuity in the pharmaceutical sector and have optimized our operations to deliver reliable performance regardless of market volatility.
We invite you to engage with our technical procurement team to discuss how this advanced synthesis can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits specific to your volume requirements. We are prepared to provide specific COA data and route feasibility assessments to support your regulatory filings and process validation efforts. Partner with us to leverage this innovative chemistry and secure a competitive advantage in the production of next-generation antiviral therapeutics.