Revolutionizing Carbetocin Manufacturing: A Deep Dive into High-Yield Solid-Phase Peptide Synthesis and Commercial Scalability
The pharmaceutical landscape for oxytocin analogues is undergoing a significant transformation driven by the urgent need for higher purity and more sustainable manufacturing processes. Patent CN106084015B introduces a groundbreaking methodology for the synthesis of carbetocin, a critical long-acting oxytocin analogue used extensively in preventing postpartum hemorrhage. This intellectual property details a refined solid-phase peptide synthesis (SPPS) strategy that fundamentally alters the production paradigm by replacing problematic liquid-phase cyclization steps with a robust on-resin approach. By leveraging a novel amino resin structure and optimizing protecting group chemistry, specifically substituting traditional Alloc groups with tBu moieties, the invention achieves a remarkable purity profile exceeding 99.5% while maintaining a total yield above 70%. For global procurement leaders and R&D directors, this patent represents a pivotal shift towards more reliable carbetocin supplier capabilities, offering a pathway to reduce supply chain risks associated with heavy metal contamination and low-yield batch processing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial preparation of carbetocin has been plagued by significant technical bottlenecks that hindered cost-effective mass production. Early methodologies, such as those described in European patent ES2115543 and Czech patent CS8605461, relied heavily on solid-liquid combined synthesis where the critical cyclization step occurred in the liquid phase. This approach inherently suffered from low cyclization yields due to the entropic penalty of bringing two distant ends of a linear peptide together in solution, often leading to intermolecular polymerization rather than the desired intramolecular ring closure. Furthermore, subsequent patents like CN101555272A attempted to address this by using palladium-catalyzed deprotection of Alloc groups; however, this introduced severe complications. The exposure of free sulfhydryl groups during deprotection frequently poisoned the expensive palladium catalysts, leading to incomplete reactions and the introduction of difficult-to-remove heavy metal impurities. These legacy processes resulted in products with single impurity levels hovering around 0.2% and total yields struggling to exceed 60%, creating substantial waste disposal challenges and inflating the cost of goods sold for manufacturers.
The Novel Approach
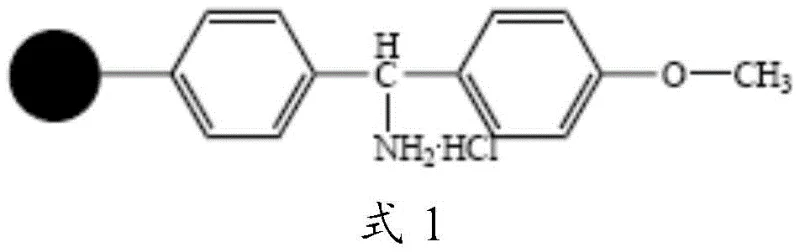
The methodology disclosed in CN106084015B circumvents these historical failures through a meticulously engineered solid-phase strategy that keeps the growing peptide chain anchored to a specialized resin throughout the critical cyclization event. By utilizing a brand-new amino resin, depicted in the structural formula below, the synthesis ensures that the reactive termini are held in close proximity, dramatically favoring intramolecular cyclization over intermolecular aggregation. Crucially, the invention replaces the problematic Alloc protecting group on the Cysteine side chain with a tert-butyl (tBu) ester. This strategic substitution eliminates the need for palladium catalysts entirely, thereby removing the risk of catalyst poisoning and the subsequent burden of heavy metal clearance. The result is a streamlined process where the maximum single impurity is reduced to less than 0.1%, and the total yield is consistently pushed above 70%, representing a quantum leap in process efficiency and product quality for high-purity pharmaceutical intermediates.
Mechanistic Insights into Solid-Phase Cyclization and tBu Deprotection
The core mechanistic advantage of this synthesis lies in the precise control of steric and electronic environments on the solid support. The process initiates with the coupling of protected Glycine to the novel MBHA-type resin, where the substitution value is optimized between 0.5 to 1.0 mmol/g to balance loading capacity with steric accessibility. As the peptide chain is extended from the C-terminus to the N-terminus using standard Fmoc or Boc chemistry, the choice of condensation reagents such as DIC/HOBt or HATU/DIPEA ensures rapid and racemization-free amide bond formation. The pivotal moment occurs after the full linear sequence is assembled; the removal of the tBu group from the Cysteine side chain using mild acidic conditions (e.g., TFA/DCM) exposes the carboxylic acid without affecting the rest of the protected peptide. This free acid is then activated on-resin to attack the N-terminal amine, closing the ring. Because the peptide is immobilized, the effective molarity of the reacting ends is exceptionally high, driving the equilibrium towards the cyclic product with minimal formation of dimers or oligomers, which are common impurities in solution-phase synthesis.
Impurity control is further enhanced by the stability of the tBu protecting group during the elongation phases. Unlike the Alloc group, which requires orthogonal deprotection conditions that can sometimes compromise acid-labile side chains, the tBu group remains inert until the specific deprotection step. This orthogonality ensures that side reactions, such as premature cyclization or aspartimide formation, are minimized. Furthermore, the final acidolysis step using HBr/TFA not only cleaves the peptide from the resin but also removes remaining acid-labile protecting groups in a single operation. This convergence of deprotection and cleavage simplifies the workflow significantly, reducing the number of unit operations and potential points of failure. The resulting crude product typically exhibits a purity of over 82%, which is exceptionally high for a crude peptide, thereby easing the load on the downstream preparative HPLC purification and significantly increasing the recovery rate of the final active pharmaceutical ingredient.
How to Synthesize Carbetocin Efficiently
The synthesis of carbetocin via this patented route requires strict adherence to reaction parameters to ensure the high yields and purity profiles described. The process involves a sequential assembly of amino acids on a specialized resin, followed by specific deprotection and cyclization events that define the quality of the final product. Operators must pay close attention to the stoichiometry of coupling reagents and the duration of deprotection steps to prevent incomplete reactions that could lead to deletion sequences. The following guide outlines the critical operational phases derived from the patent examples, serving as a foundational reference for process chemists aiming to replicate this high-efficiency route in a GMP environment.
- Couple protected Glycine to the novel amino resin (Formula 1) using condensation reagents like DIC and HOBt to form Peptide Resin 1.
- Perform sequential extension coupling from the C-terminus to the N-terminus using protected amino acids (Leu, Pro, Cys(tBu), Asn, Gln, Ile, Tyr) to build the peptide chain.
- Remove the tBu protecting group from the Cysteine side chain to expose the carboxyl group for subsequent cyclization.
- Execute intramolecular coupling cyclization on the resin to form the cyclic peptide structure characteristic of carbetocin.
- Cleave the peptide from the resin using an acidolysis agent such as HBr/TFA solution to obtain the crude carbetocin product.
- Purify the crude product via preparative HPLC and perform salt conversion to achieve final purity exceeding 99.5%.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthesis route offers profound strategic benefits that extend far beyond simple chemical yield improvements. The elimination of palladium catalysts, which are subject to volatile market pricing and supply constraints, immediately stabilizes the raw material cost structure. Moreover, the removal of heavy metals from the process flow drastically reduces the complexity and cost of wastewater treatment and regulatory compliance, as there is no longer a need for specialized scavenging resins or extensive testing for residual metals. This simplification translates directly into a more resilient supply chain, where production batches are less likely to be delayed by purification bottlenecks or failed quality control tests related to metal content. The robustness of the solid-phase method also implies a higher success rate per batch, ensuring consistent availability of this critical uterotonic agent for global markets.
- Cost Reduction in Manufacturing: The transition to a tBu-based protection strategy eliminates the requirement for expensive transition metal catalysts and their associated ligands, which traditionally accounted for a significant portion of reagent costs. By avoiding these costly inputs and the subsequent purification steps required to remove them, the overall cost of goods sold is substantially decreased. Additionally, the higher total yield exceeding 70% means that less starting material is wasted per kilogram of final product, further driving down the unit cost and improving margin potential for large-scale manufacturers.
- Enhanced Supply Chain Reliability: Dependence on specialized palladium catalysts often introduces single points of failure in the supply chain, as these materials can face geopolitical or logistical disruptions. By shifting to a purely organic synthesis pathway using widely available acidolysis agents and standard coupling reagents, the manufacturing process becomes far more robust against external shocks. This reliability ensures that production schedules can be maintained with greater certainty, reducing lead times for high-purity pharmaceutical intermediates and guaranteeing continuity of supply for downstream drug product manufacturers.
- Scalability and Environmental Compliance: The solid-phase nature of this synthesis is inherently scalable, allowing for seamless transition from laboratory benchtop quantities to multi-ton commercial production without fundamental changes to the chemistry. The reduction in hazardous heavy metal waste aligns perfectly with increasingly stringent environmental regulations, minimizing the ecological footprint of the manufacturing facility. This environmental compliance not only mitigates regulatory risk but also enhances the corporate sustainability profile, a key metric for modern pharmaceutical partnerships.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis method. These answers are derived directly from the experimental data and technical disclosures within the patent documentation, providing clarity on how this technology compares to incumbent processes. Understanding these nuances is essential for technical teams evaluating the feasibility of technology transfer or licensing agreements.
Q: How does the novel solid-phase method improve carbetocin purity compared to traditional liquid-phase cyclization?
A: Traditional liquid-phase methods often suffer from low cyclization yields and complex purification due to intermolecular polymerization. The novel solid-phase method described in patent CN106084015B utilizes a specific MBHA resin and tBu protection strategy, which confines the reaction to the solid support, effectively preventing intermolecular side reactions and achieving a purity of over 99.5% with single impurities below 0.1%.
Q: What are the advantages of using tBu protection over Alloc protection for the Cysteine side chain in this synthesis?
A: Previous methods utilizing Alloc protection required palladium catalysts (e.g., tetrakis(triphenylphosphine)palladium) for deprotection, which introduced heavy metal contaminants and risked catalyst poisoning by exposed sulfhydryl groups. The new method employs tBu protection, which is removed under acidic conditions without heavy metals, thereby simplifying purification, reducing toxic waste, and eliminating the risk of catalyst deactivation.
Q: What total yield can be expected from this optimized synthetic route?
A: While earlier patents reported total yields as low as 51% due to improper protecting group selection and purification losses, this optimized protocol demonstrates a robust total yield exceeding 70%. This significant improvement is attributed to the high loading capacity of the novel resin and the efficiency of the tBu deprotection and cyclization steps.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Carbetocin Supplier
The technical advancements detailed in patent CN106084015B underscore the immense potential for producing carbetocin with unprecedented purity and efficiency. At NINGBO INNO PHARMCHEM, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring such sophisticated peptide synthesis routes to life. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, ensuring that every batch of carbetocin we produce adheres to the highest international standards for pharmaceutical intermediates. We understand that consistency is key in the pharma industry, and our process engineering teams are dedicated to maintaining the delicate balance of reaction conditions necessary to achieve the >99.5% purity benchmarks outlined in this innovative patent.
We invite forward-thinking partners to collaborate with us to leverage this superior synthesis technology for your supply chain needs. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis that quantifies the specific economic benefits of switching to this heavy-metal-free process for your organization. We encourage you to contact us today to discuss specific COA data and route feasibility assessments, ensuring that your next project is built on a foundation of chemical excellence and supply chain security.