Advanced Asymmetric Synthesis of Chiral Statin Intermediates for Commercial Scale Production
Advanced Asymmetric Synthesis of Chiral Statin Intermediates for Commercial Scale Production
The pharmaceutical industry's relentless pursuit of potent HMG-CoA reductase inhibitors, commonly known as statins, has driven significant innovation in synthetic organic chemistry. Patent CN102212081B introduces a groundbreaking preparation method for a critical chiral intermediate, specifically the compound of general Formula 1, which serves as a pivotal side chain for superstatins like Rosuvastatin and Pitavastatin. This technology represents a paradigm shift from traditional racemic resolution strategies to a more efficient asymmetric synthesis approach. By leveraging S-benzyl mandelate as a chiral source, the process effectively installs the required (3R) stereochemistry early in the synthesis, thereby circumventing the inherent 50% yield loss associated with resolving racemic mixtures. This advancement is crucial for manufacturers seeking to optimize production costs while maintaining the stringent optical purity standards required for regulatory approval in global markets.
For procurement specialists and supply chain directors, the implications of this synthetic route are profound. The ability to produce high-purity statin intermediates without the waste generation inherent in resolution processes translates directly to improved sustainability metrics and reduced raw material consumption. As a reliable statin intermediate supplier, understanding the nuances of such proprietary pathways allows us to offer clients not just a commodity, but a strategic advantage in their drug development pipelines. The structural complexity of statin molecules, featuring multiple chiral centers and sensitive functional groups, demands a synthesis strategy that balances reactivity with selectivity. This patent addresses those challenges head-on, providing a robust framework for the commercial scale-up of complex pharmaceutical intermediates that meets the rigorous demands of modern drug manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of statin side chains has relied heavily on methods that involve the resolution of racemic precursors, a strategy fraught with inefficiency. For instance, prior art such as Document WO2008130678 describes a route where a racemic mono-ester is subjected to chiral resolution using phenylethylamine. While chemically feasible, this approach fundamentally caps the theoretical yield at 50%, as the unwanted enantiomer is typically discarded or requires energy-intensive recycling. Furthermore, the use of methyl esters as protecting groups in some conventional routes poses stability risks; under basic deprotection conditions, methyl esters can facilitate lactone formation, leading to racemization of the sensitive 5-hydroxyl position. This side reaction compromises the optical purity of the final API, necessitating costly and time-consuming purification steps that erode profit margins.
In addition to yield losses, conventional methods often employ Wittig or Wittig-Horner reactions that require harsh conditions or generate stoichiometric amounts of phosphine oxide waste, complicating downstream processing. The reliance on resolution means that manufacturers must process double the amount of starting material to achieve the same output of active chiral material, significantly inflating the cost of goods sold (COGS). For a procurement manager focused on cost reduction in pharmaceutical manufacturing, these inefficiencies represent a substantial burden. The accumulation of impurities from side reactions and the need for multiple crystallization steps to upgrade enantiomeric excess further extend lead times, creating bottlenecks in the supply chain that can delay critical clinical trials or commercial launches.
The Novel Approach
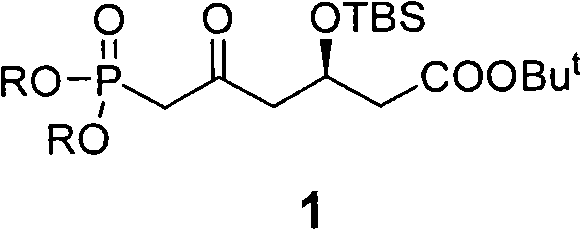
The methodology disclosed in CN102212081B offers a sophisticated solution by employing an asymmetric induction strategy that bypasses the need for resolution entirely. The core innovation lies in the initial coupling of 3-(tert-butyldimethylsilyl)-pyroglutaric acid with S-benzyl mandelate. This reaction, facilitated by a strong organometallic base like n-Butyl Lithium at cryogenic temperatures (-78°C), establishes the chiral center with high fidelity from the outset. By utilizing a tert-butyl ester for carboxyl protection instead of a methyl ester, the process enhances stability against base-catalyzed racemization, ensuring that the hard-won optical purity is preserved throughout the subsequent synthetic transformations. This strategic choice of protecting groups is a testament to the process's design for robustness and scalability.
Moreover, the novel route streamlines the synthesis into a logical sequence of seven steps that utilize standard, commercially available reagents. The elimination of the resolution step not only doubles the potential yield relative to racemic starting materials but also simplifies the operational workflow. There is no need for the complex separation of diastereomeric salts or the handling of large volumes of mother liquors containing the unwanted enantiomer. This streamlined approach significantly reduces the environmental footprint of the manufacturing process, aligning with the growing industry emphasis on green chemistry principles. For supply chain heads, this translates to a more predictable and reliable supply of high-purity intermediates, reducing the risk of stockouts and ensuring continuity of supply for downstream API production.
Mechanistic Insights into Asymmetric Induction and Phosphonate Coupling
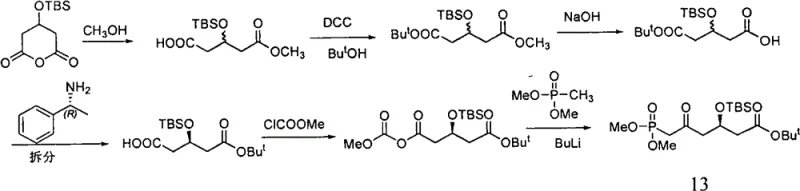
The success of this synthesis hinges on the precise control of stereochemistry during the initial nucleophilic attack. In the first step, the deprotonated S-benzyl mandelate acts as a chiral nucleophile, attacking the carbonyl of the silyl-protected pyroglutaric acid derivative. The reaction is conducted in tetrahydrofuran (THF) at -78°C to minimize non-selective background reactions and maximize the diastereoselectivity of the addition. The bulky tert-butyldimethylsilyl (TBS) group plays a dual role here: it protects the hydroxyl functionality from unwanted side reactions and provides steric bulk that aids in directing the approach of the nucleophile. Following this key bond-forming event, the benzyl group is removed via catalytic hydrogenation using Pd/C. This mild deprotection method is crucial as it avoids the use of strong acids or bases that could epimerize the newly formed chiral center, preserving the optical purity which is reported to exceed 99.5% ee after recrystallization.
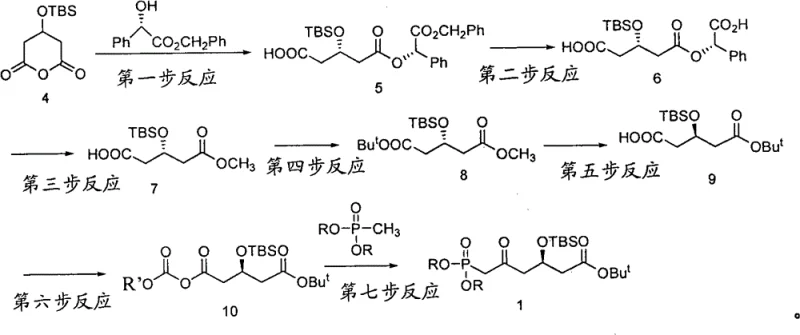
The latter stages of the synthesis involve the installation of the phosphonate moiety, which is essential for the subsequent Wittig-Horner coupling with the statin lactone ring. The process converts the carboxylic acid into a mixed anhydride using methyl chloroformate at low temperatures (-60°C), activating it for nucleophilic attack by the lithiated dialkyl methyl phosphonate. This acylation of the phosphonate carbanion is a delicate operation that requires strict temperature control to prevent decomposition of the reactive anion. The use of a tert-butyl ester at the other end of the molecule ensures orthogonality; it remains intact during the basic conditions of the phosphonate addition but can be selectively removed later under acidic conditions if necessary. This careful orchestration of protecting group chemistry and reaction conditions ensures that the final intermediate possesses the exact structural and stereochemical attributes required for high-yielding coupling in the final API synthesis.
How to Synthesize Chiral Statin Side Chain Efficiently
The synthesis of this high-value intermediate requires meticulous attention to reaction parameters, particularly temperature and stoichiometry, to ensure consistent quality. The process begins with the preparation of the chiral diester, followed by a sequence of deprotection, esterification, and functional group interconversion. Each step has been optimized in the patent examples to provide clear guidance on reagent equivalents and workup procedures. For example, the hydrogenation step specifies the use of ethanol as a solvent and normal pressure hydrogen, making it easily adaptable to standard plant equipment. The detailed experimental data provided in the patent, including NMR characterization and melting points, serves as a vital benchmark for quality control teams aiming to replicate this process. Detailed standardized synthesis steps are provided in the guide below to assist technical teams in implementation.
- React 3-(tert-butyldimethylsilyl)-pyroglutaric acid with S-benzyl mandelate using n-Butyl Lithium at -78°C to form the chiral diester intermediate.
- Perform catalytic hydrogenation to remove the benzyl protecting group, followed by transesterification with sodium methylate to obtain the monomethyl glutarate.
- Condense with tert-butanol using DCC/DMAP, hydrolyze the methyl ester, activate with methyl chloroformate, and finally react with dialkyl methyl phosphonate to yield the target phosphonate.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting this advanced synthetic route offers tangible benefits that extend far beyond the laboratory bench, directly impacting the bottom line and operational resilience of pharmaceutical manufacturing. By shifting from a resolution-based process to an asymmetric synthesis, companies can fundamentally alter their cost structure. The elimination of the 50% yield loss inherent in resolution means that less raw material is required to produce the same amount of active intermediate. This efficiency gain is compounded by the reduction in solvent usage and waste disposal costs, as there are no mother liquors rich in the unwanted enantiomer to treat. Furthermore, the use of stable tert-butyl protecting groups reduces the incidence of batch failures due to racemization, leading to higher overall process reliability and fewer costly reworks.
- Cost Reduction in Manufacturing: The primary economic driver of this technology is the drastic improvement in atom economy and yield efficiency. By avoiding the discard of half the starting material, the effective cost per kilogram of the chiral intermediate is significantly lowered. Additionally, the process avoids the use of expensive chiral catalysts or enzymes often required in biocatalytic resolutions, relying instead on cost-effective chiral pool materials like S-benzyl mandelate. The simplified purification profile, characterized by crystallization rather than column chromatography, further reduces operational expenditures related to silica gel and solvent recovery, making the process economically viable for large-scale production.
- Enhanced Supply Chain Reliability: From a sourcing perspective, the reagents used in this pathway—such as pyroglutaric acid derivatives, DCC, and standard phosphonates—are commodity chemicals with robust global supply chains. This reduces the risk of supply disruptions that can occur with specialized chiral catalysts or bespoke reagents. The robustness of the chemistry, demonstrated by high yields in the patent examples (e.g., 96.6% for transesterification, 86.1% for hydrolysis), suggests a process that is forgiving of minor variations in scale-up, ensuring consistent delivery schedules. This reliability is critical for maintaining the continuous flow of materials required for just-in-time API manufacturing.
- Scalability and Environmental Compliance: The process is designed with scale-up in mind, utilizing unit operations that are standard in the fine chemical industry, such as low-temperature batch reactions and filtration. The avoidance of heavy metal catalysts (other than recyclable Pd/C) and the minimization of hazardous waste streams align with increasingly stringent environmental regulations. This 'green' profile not only reduces compliance costs but also enhances the corporate social responsibility standing of the manufacturer. The ability to scale from 100 kgs to 100 MT annual commercial production without fundamental changes to the chemistry ensures that the supply can grow in lockstep with the market demand for statin therapies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the patent specifications and are intended to clarify the operational advantages and chemical rationale behind the process. Understanding these details is essential for technical teams evaluating the feasibility of adopting this route for their specific manufacturing needs. The answers reflect the balance between high-performance chemistry and practical industrial application.
Q: Why is this asymmetric method superior to traditional resolution methods?
A: Traditional methods often rely on resolving racemic mixtures, which theoretically limits maximum yield to 50% and requires complex separation steps. This patented asymmetric synthesis utilizes a chiral auxiliary (S-benzyl mandelate) to induce stereochemistry directly, potentially doubling theoretical yield and simplifying purification.
Q: What are the critical reaction conditions for maintaining optical purity?
A: The initial nucleophilic addition must be conducted at low temperatures, specifically between -60°C to -100°C (preferably -78°C), to ensure high stereoselectivity. Additionally, the hydrogenation step uses Pd/C under normal pressure to gently remove the benzyl group without affecting the chiral center.
Q: Is this process suitable for large-scale industrial manufacturing?
A: Yes, the process avoids chromatography and uses standard reagents like DCC, n-Butyl Lithium, and common solvents (THF, Toluene, Ethanol). The elimination of resolution steps and the use of robust protection groups (TBS, t-Butyl) make it highly amenable to scale-up from kilograms to metric tons.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Statin Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of life-saving medications depends on the availability of high-quality, cost-effective intermediates. Our team of expert chemists has extensively analyzed the pathway described in CN102212081B and possesses the technical capability to execute this complex asymmetric synthesis with precision. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and reliability. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, including the critical enantiomeric excess required for statin side chains, guaranteeing that every batch meets global regulatory standards.
We invite you to collaborate with us to leverage this advanced technology for your statin projects. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis that quantifies the potential economic benefits of switching to this resolution-free route for your specific volume requirements. Please contact us to request specific COA data from our pilot batches and comprehensive route feasibility assessments. Let us partner with you to optimize your supply chain and accelerate the delivery of affordable cardiovascular therapies to patients worldwide.