Scalable Synthesis of Ethyl (R)-4-Cyano-3-Hydroxybutyrate for Commercial Atorvastatin Production
The global demand for statins, particularly Atorvastatin Calcium, continues to drive significant innovation in the synthesis of key pharmaceutical intermediates. Patent CN102442927B introduces a robust and highly efficient preparation method for Ethyl (R)-(-)-4-cyano-3-hydroxybutyrate, a critical chiral building block in the Atorvastatin value chain. This technology addresses long-standing challenges in stereochemical control and yield optimization that have historically plagued the manufacturing of this specific intermediate. By shifting away from direct substitution methods that suffer from competing side reactions, this novel approach utilizes a strategic epoxide intermediate to ensure high fidelity in chirality transfer. For R&D directors and process chemists, this represents a pivotal shift towards more predictable and cleaner synthetic routes. The methodology not only enhances the overall mass balance of the production line but also aligns with modern green chemistry principles by reducing the reliance on complex purification techniques such as column chromatography. As the pharmaceutical industry seeks to optimize cost structures while maintaining rigorous quality standards, the adoption of such refined synthetic pathways becomes essential for maintaining competitive advantage in the generic and branded drug markets.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Ethyl (R)-4-cyano-3-hydroxybutyrate has been fraught with significant technical and economic hurdles that limit industrial viability. One conventional pathway relies on the use of Ethyl (S)-(-)-4-bromo-3-hydroxybutyrate as the starting material; while this route offers reasonable reactivity due to the good leaving group ability of bromine, the raw material itself is prohibitively expensive and difficult to source in bulk quantities, rendering it unsuitable for cost-sensitive commercial manufacturing. Another prevalent method involves the direct nucleophilic substitution of Ethyl (S)-(-)-4-chloro-3-hydroxybutyrate with sodium cyanide under alkaline conditions. However, this direct approach is chemically inefficient because the basic environment required for cyanation simultaneously promotes undesirable elimination reactions, leading to the formation of olefinic byproducts, as well as hydrolysis reactions that degrade the ester functionality. These competing pathways drastically reduce the isolated yield of the desired nitrile, often capping efficiency at merely 40% to 50% after purification, which creates substantial waste disposal issues and inflates the cost of goods sold. Furthermore, older methods often necessitated the use of trimethylsilyl protecting groups to mask the hydroxyl functionality, introducing additional synthetic steps, expensive reagents, and moisture sensitivity that complicate scale-up operations in standard stainless steel reactors.
The Novel Approach
In stark contrast to these legacy methods, the technology disclosed in CN102442927B employs a sophisticated two-step sequence that decouples the activation of the leaving group from the introduction of the cyano moiety, thereby circumventing the harsh conditions that cause degradation. The process initiates with an intramolecular cyclization of the chloro-hydroxy precursor to form Ethyl 3,4-epoxybutyrate, a stable and versatile intermediate that locks the stereochemical information without exposing the molecule to strong nucleophiles prematurely. This epoxidation step is conducted under mild alkaline conditions using inexpensive solid bases and catalytic amounts of silver salts, ensuring high conversion rates without the aggressive thermal stress associated with direct cyanation. Subsequently, the epoxide ring is opened regioselectively using a cyanide source in a controlled acidic environment, which directs the nucleophilic attack to the correct carbon atom to establish the desired (R)-configuration with high optical purity. This strategic detour through the epoxide intermediate effectively suppresses the elimination and hydrolysis side reactions that plague direct substitution, resulting in a dramatic improvement in overall yield, consistently achieving between 85% and 95% across multiple experimental batches. The final product is isolated via vacuum distillation rather than chromatography, a critical modification that transforms the process from a laboratory curiosity into a viable industrial operation capable of multi-ton production.
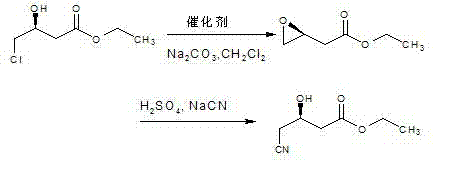
Mechanistic Insights into Epoxide-Mediated Stereocontrol
The core chemical innovation of this process lies in the precise manipulation of stereochemistry through the epoxide intermediate, which serves as a rigid conformational lock during the transformation. In the first stage, the treatment of Ethyl (S)-4-chloro-3-hydroxybutyrate with a solid base facilitates the deprotonation of the secondary hydroxyl group, generating an alkoxide species that immediately undergoes an intramolecular SN2 attack on the adjacent carbon bearing the chlorine atom. This internal displacement results in the formation of the three-membered oxirane ring with inversion of configuration at the carbon center, effectively storing the chiral information in a strained but stable cyclic ether. The use of a silver-based catalyst in this step is particularly noteworthy, as it likely assists in the abstraction of the chloride ion, lowering the activation energy for ring closure and allowing the reaction to proceed efficiently at temperatures ranging from 0°C to 50°C. This mild thermal profile is crucial for preventing the thermal decomposition of the sensitive ester group and minimizing racemization, ensuring that the optical integrity of the starting material is preserved throughout the cyclization event. The resulting Ethyl 3,4-epoxybutyrate is isolated with high purity, providing a clean substrate for the subsequent functionalization step.
The second mechanistic phase involves the regioselective ring-opening of the epoxide by the cyanide anion, which is the determinant step for establishing the final (R)-configuration of the product. Under the controlled pH conditions of 8 to 10 maintained by the simultaneous addition of acid and cyanide salt, the nucleophilic cyanide ion attacks the less sterically hindered terminal carbon of the epoxide ring. This attack occurs with inversion of configuration at the site of substitution, but due to the specific geometry of the epoxide formed in the previous step, the net result is the retention of the desired relative stereochemistry required for the Atorvastatin side chain. The acidic environment is carefully tuned to protonate the resulting alkoxide intermediate without causing hydrolysis of the newly formed nitrile group or the existing ester moiety. This delicate balance of pH and temperature (-10°C to 30°C) ensures that the reaction proceeds with high chemoselectivity, avoiding the formation of diol byproducts that would occur under strongly acidic or basic conditions. The mechanism effectively isolates the reactive centers, allowing for a clean transformation that is far superior to the chaotic mixture of products observed in direct displacement reactions, ultimately delivering a product with purity levels exceeding 99% as confirmed by chiral HPLC and NMR analysis.
How to Synthesize Ethyl (R)-4-Cyano-3-Hydroxybutyrate Efficiently
The operational protocol for this synthesis is designed to maximize throughput while minimizing the complexity of workup procedures, making it ideally suited for pilot plant and commercial scale implementation. The process begins with the dissolution of the chloro-hydroxy starting material in a common organic solvent such as dichloromethane or acetonitrile, followed by the addition of a stoichiometric amount of solid base like sodium carbonate or potassium carbonate. The reaction mixture is stirred at ambient or slightly elevated temperatures until gas chromatography indicates complete consumption of the starting material, typically requiring between 3 to 48 hours depending on the specific solvent and base combination employed. Once the epoxide intermediate is formed, the solvent is removed, and the crude epoxide is subjected to the ring-opening conditions using an aqueous cyanide solution buffered with sulfuric or citric acid. The detailed standardized synthesis steps, including specific molar ratios, agitation speeds, and quenching procedures, are outlined in the structured guide below to ensure reproducibility and safety during handling of cyanide reagents.
- Cyclization: Dissolve ethyl (S)-4-chloro-3-hydroxybutyrate in organic solvent, add solid base and catalyst at 0-50°C to form ethyl 3,4-epoxybutyrate.
- Ring-Opening: React the epoxide intermediate with cyanide reagent (NaCN/KCN) and acid at pH 8-10 and -10 to 30°C.
- Purification: Extract with organic solvent, recover layer, and purify via vacuum distillation to achieve >99% purity.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this epoxide-mediated synthesis route offers transformative advantages that directly impact the bottom line and operational resilience of pharmaceutical manufacturing. The primary economic driver is the substitution of expensive and scarce bromo-precursors with the widely available and cost-effective chloro-analog, which significantly reduces the raw material cost basis without compromising reaction kinetics. Furthermore, the elimination of protective group chemistry, specifically the removal of trimethylsilyl reagents, simplifies the bill of materials and reduces the dependency on specialty chemical suppliers who may have volatile pricing or long lead times. The robustness of the reaction conditions, which tolerate a broader range of temperatures and pH levels compared to direct cyanation, translates into higher process reliability and reduced batch failure rates, ensuring a consistent supply of critical intermediates for downstream API production. Additionally, the ability to purify the final product via vacuum distillation rather than silica gel chromatography represents a massive reduction in processing time and solvent consumption, facilitating faster turnaround times from reactor to warehouse and enabling true continuous manufacturing potential.
- Cost Reduction in Manufacturing: The economic benefits of this process are derived principally from the valorization of low-cost starting materials and the minimization of waste generation through high-yield transformations. By avoiding the use of premium-priced bromo-compounds and expensive silylating agents, the direct material costs are substantially lowered, allowing for more competitive pricing in the generic pharmaceutical market. The high selectivity of the epoxide ring-opening step means that fewer byproducts are formed, which reduces the load on wastewater treatment facilities and lowers the cost associated with hazardous waste disposal. Moreover, the replacement of chromatographic purification with distillation allows for the recovery and recycling of solvents, further driving down the variable costs per kilogram of product. This lean manufacturing approach ensures that the cost of goods sold remains optimized even as regulatory requirements for purity become more stringent, providing a sustainable margin structure for long-term production contracts.
- Enhanced Supply Chain Reliability: Supply chain security is significantly bolstered by the use of commodity chemicals that are readily available from multiple global suppliers, reducing the risk of single-source bottlenecks. The starting material, Ethyl (S)-4-chloro-3-hydroxybutyrate, is a mature industrial chemical with a stable supply network, unlike the niche bromo-derivatives which can be subject to availability fluctuations. The process tolerance for various solid bases and acids provides procurement teams with the flexibility to switch vendors based on price and delivery schedules without needing to requalify the entire process. The simplified workflow, which eliminates complex protection-deprotection sequences, shortens the overall cycle time from raw material intake to finished goods, thereby improving inventory turnover rates. This agility allows manufacturers to respond more rapidly to spikes in demand for Atorvastatin, ensuring that downstream formulation lines are never starved of critical intermediates due to upstream synthesis delays.
- Scalability and Environmental Compliance: The transition from laboratory to commercial scale is seamless with this methodology due to the reliance on standard unit operations that are well-understood in chemical engineering. The exothermic nature of the reactions is manageable within standard jacketed reactors, and the absence of pyrophoric or highly unstable reagents simplifies safety protocols and insurance requirements. From an environmental standpoint, the high atom economy of the cyclization and ring-opening steps minimizes the E-factor (mass of waste per mass of product), aligning with increasingly strict global environmental regulations. The use of distillation for purification avoids the generation of tons of spent silica gel waste, which is a significant environmental burden in traditional fine chemical synthesis. This green chemistry profile not only reduces compliance costs but also enhances the corporate sustainability metrics of the manufacturing entity, making it a preferred partner for environmentally conscious multinational pharmaceutical companies seeking to reduce their carbon footprint.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology, drawing directly from the experimental data and process parameters defined in the patent literature. Understanding these nuances is critical for process engineers and quality assurance teams who are evaluating the feasibility of integrating this route into existing production facilities. The answers provided reflect the specific advantages of the epoxide intermediate strategy over traditional methods, highlighting the improvements in yield, purity, and operational safety that define this modern approach to Atorvastatin intermediate manufacturing.
Q: Why is the epoxide intermediate route superior to direct cyanation?
A: Direct cyanation of the chloro-precursor often leads to elimination and hydrolysis side reactions, resulting in low yields (~40%). The epoxide route minimizes these impurities, achieving yields of 85-95%.
Q: What is the expected purity of the final product using this method?
A: The patented process utilizes vacuum distillation rather than column chromatography, consistently delivering product purity exceeding 99%, which is critical for API synthesis.
Q: Is this process suitable for large-scale industrial manufacturing?
A: Yes, the method avoids expensive reagents like silyl protecting groups or bromo-precursors and uses standard unit operations like filtration and distillation, making it highly scalable.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ethyl (R)-4-Cyano-3-Hydroxybutyrate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of life-saving medications like Atorvastatin depends on the unwavering quality and availability of key chiral intermediates. Our technical team has extensively analyzed the pathway described in CN102442927B and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this efficient synthesis to life. We operate state-of-the-art facilities equipped with rigorous QC labs capable of verifying stringent purity specifications, ensuring that every batch of Ethyl (R)-4-Cyano-3-Hydroxybutyrate meets the exacting standards required for GMP API synthesis. Our commitment to process excellence means that we can deliver this critical intermediate with the consistency and reliability that global supply chains demand, mitigating the risks associated with complex multi-step syntheses.
We invite procurement leaders and R&D directors to engage with us to discuss how this optimized synthetic route can enhance your project economics and timeline. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis that quantifies the potential reductions in raw material and waste disposal costs specific to your volume requirements. We encourage you to contact us directly to obtain specific COA data from our recent pilot runs and to receive detailed route feasibility assessments tailored to your manufacturing capabilities. Let us be your strategic partner in securing a robust and cost-effective supply of high-purity pharmaceutical intermediates for the future.