Advanced Semi-Synthetic Route for High-Purity Orlistat: Technical Analysis and Commercial Scalability
Advanced Semi-Synthetic Route for High-Purity Orlistat: Technical Analysis and Commercial Scalability
The pharmaceutical industry constantly seeks robust manufacturing pathways that balance high purity with economic viability, particularly for high-demand anti-obesity agents. Patent CN102304105B introduces a groundbreaking method for preparing high-purity Orlistat, leveraging a semi-synthetic approach that starts from Lipstatin fermentation broth. This technology addresses critical bottlenecks in existing production methods by integrating mixed silica gel medium-pressure chromatography with precise crystallization techniques. Unlike traditional total synthesis routes that suffer from excessive step counts and low overall yields, this innovation achieves a total yield greater than 30% while maintaining product purity above 99.5% with single impurities controlled below 0.1%. For R&D directors and procurement specialists, this represents a significant opportunity to secure a reliable pharmaceutical intermediate supplier capable of delivering material that strictly adheres to ICH guidelines without the prohibitive costs associated with preparative HPLC purification.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the production of Orlistat has been plagued by significant technical and economic challenges that hinder efficient commercial scale-up of complex pharmaceutical intermediates. Traditional total synthesis methods, such as those disclosed in earlier patents, often require up to twelve distinct reaction steps, resulting in cumulative yield losses and exorbitant reagent costs. Furthermore, purification strategies relying on Dynamic Axial Compression (DAC) or reverse-phase high-performance liquid chromatography, while capable of achieving high purity, demand substantial capital investment in specialized equipment and expensive filler materials. These methods also typically require the crude material to have a pre-purification purity exceeding 90%, creating a stringent upstream bottleneck. Additionally, batch processing capacities in these conventional systems are often limited, making them unsuitable for the multi-ton production scales required to meet global market demand effectively.
The Novel Approach
The methodology outlined in patent CN102304105B fundamentally restructures the production workflow to overcome these legacy inefficiencies through a clever combination of semi-synthesis and optimized downstream processing. By utilizing Lipstatin derived directly from Streptomyces toxytricini fermentation, the process bypasses the lengthy construction of the core beta-lactone structure, thereby drastically simplifying the synthetic route. The core innovation lies in the deployment of mixed silica gel medium-pressure chromatography, which utilizes specific mesh combinations (such as 100-200 mesh and 200-300 mesh) to achieve superior separation resolution at a fraction of the cost of DAC systems. This is followed by a rigorous sequence of decolorization, catalytic hydrogenation, and multi-stage crystallization. This holistic approach not only ensures the final product meets the stringent purity specifications of over 99.5% but also utilizes standard chemical engineering equipment, significantly lowering the barrier to entry for cost reduction in API manufacturing.
Mechanistic Insights into Catalytic Hydrogenation and Chromatographic Separation
The chemical transformation at the heart of this process is the catalytic hydrogenation of Lipstatin to Orlistat, a reaction that requires precise control to prevent degradation of the sensitive beta-lactone ring. As illustrated in the reaction scheme below, the process involves the saturation of specific double bonds in the Lipstatin side chain using a palladium on carbon (Pd/C) catalyst under controlled hydrogen pressure. The patent specifies operating pressures between 0.1 MPa and 1.0 MPa and temperatures ranging from 20°C to 50°C. Maintaining these parameters is critical; excessive pressure or temperature could lead to over-reduction or ring-opening side reactions, which would generate impurities difficult to remove in subsequent steps. The use of Pd/C allows for heterogeneous catalysis, facilitating easy filtration and catalyst recovery, which is essential for minimizing heavy metal residues in the final active pharmaceutical ingredient.
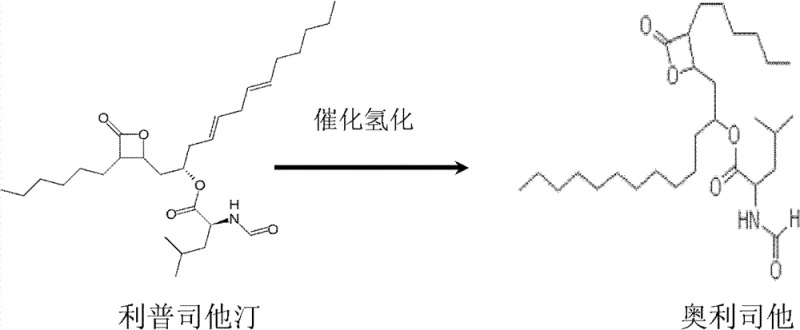
Beyond the hydrogenation step, the purification mechanism relies heavily on the physicochemical properties of the stationary phase in the chromatography column. The patent details a sophisticated packing strategy where the silica gel column is loaded with a gradient of mesh sizes, specifically placing coarser silica (e.g., 200-300 mesh) at the bottom and finer silica (e.g., 100-200 mesh) at the top. This configuration optimizes flow dynamics and surface area contact, allowing for the effective separation of Lipstatin from structurally similar fermentation byproducts. The mobile phase, a carefully tuned mixture of acetone and ethyl acetate (optimized at a volume ratio of 10:90), interacts differentially with the analytes based on polarity. This precise control over the stationary and mobile phases ensures that the collected Lipstatin fraction has a purity greater than 93% before it even enters the hydrogenation stage, thereby reducing the burden on the final crystallization steps to remove trace impurities.
How to Synthesize High-Purity Orlistat Efficiently
Implementing this synthesis route requires strict adherence to the optimized parameters defined in the patent to ensure reproducibility and quality. The process begins with the extraction of Lipstatin from fermentation broth using solvents like ethanol, followed by concentration and loading onto the prepared silica column. Following chromatographic purification and decolorization with activated carbon, the material undergoes the critical hydrogenation step. The resulting crude Orlistat is then subjected to a multi-step crystallization protocol involving cooling and anti-solvent addition (purified water) to precipitate the product. Detailed standardized synthesis steps are provided in the guide below to assist technical teams in replicating this high-efficiency pathway.
- Extract Lipstatin from fermentation broth using organic solvents like ethanol, followed by liquid-liquid extraction with heptane to concentrate the active ingredient.
- Purify the concentrated Lipstatin using mixed silica gel medium-pressure chromatography with optimized mesh sizes and pressure controls to remove impurities.
- Perform catalytic hydrogenation using Pd/C to convert Lipstatin to Orlistat, followed by multi-step crystallization and crystal form conversion to ensure >99.5% purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this manufacturing technology offers profound strategic benefits that extend beyond simple unit cost metrics. The primary advantage lies in the drastic simplification of the capital expenditure profile; because the process utilizes common chemical equipment such as plate-and-frame filter presses and standard stainless steel reactors, the initial investment is significantly lower compared to facilities requiring specialized preparative HPLC systems. This lower barrier to entry enhances supply chain resilience by enabling a broader base of qualified manufacturers to produce the material, thereby reducing the risk of single-source dependency. Furthermore, the ability to recycle solvents such as ethyl acetate and ethanol creates a closed-loop system that minimizes raw material consumption and waste disposal costs, contributing to substantial long-term operational savings.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts in favor of recoverable Pd/C, combined with the use of standard silica gel instead of costly chromatography resins, drives down the variable cost of goods sold. The process design inherently supports solvent recovery, meaning that the volume of fresh solvent required per kilogram of product is minimized. This efficiency translates directly into a more competitive pricing structure for the final API, allowing pharmaceutical companies to maintain healthy margins even in price-sensitive markets. Additionally, the high total yield of over 30% ensures that the starting fermentation broth is utilized with maximum efficiency, reducing the cost burden associated with raw material sourcing.
- Enhanced Supply Chain Reliability: The reliance on readily available industrial chemicals and standard equipment means that the supply chain is less vulnerable to disruptions caused by specialized component shortages. The scalability of the process, demonstrated by production capacities reaching 1.5 to 3 tons per month in the patent examples, assures buyers that suppliers can ramp up volume quickly to meet surges in market demand. The robustness of the purification steps also ensures consistent quality output, reducing the likelihood of batch failures that could interrupt the supply of finished dosage forms to patients. This reliability is crucial for maintaining uninterrupted production schedules for weight management medications.
- Scalability and Environmental Compliance: From an environmental perspective, the process aligns well with modern green chemistry initiatives. The ability to recycle solvents significantly reduces the facility's environmental footprint and lowers the costs associated with hazardous waste treatment. The use of non-toxic solvents like ethanol and ethyl acetate further simplifies regulatory compliance regarding worker safety and emissions. As global regulations on pharmaceutical manufacturing become increasingly stringent, adopting a process that inherently minimizes pollution and maximizes resource efficiency provides a distinct competitive advantage. This sustainability profile is increasingly becoming a key criterion for procurement decisions among top-tier pharmaceutical companies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of Orlistat using this patented methodology. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing a factual basis for evaluating the technology's viability. Understanding these details is essential for stakeholders assessing the risk and potential of integrating this supply source into their portfolio.
Q: How does this method improve Orlistat purity compared to traditional synthesis?
A: Unlike total synthesis which involves over 12 steps with low yields, this semi-synthetic route utilizes fermentation-derived Lipstatin. The key innovation is the use of mixed silica gel medium-pressure chromatography combined with specific crystallization protocols, achieving purity above 99.5% and single impurities below 0.1%, meeting strict ICH guidelines without the high capital expenditure of preparative HPLC.
Q: Is this process scalable for industrial production?
A: Yes, the process is designed for scalability. It utilizes common chemical equipment such as plate-and-frame filter presses and standard hydrogenation kettles rather than expensive specialized columns. The patent demonstrates a production capacity of 1.5 to 3 tons per month with a total yield exceeding 30%, indicating robust feasibility for commercial scale-up.
Q: What are the environmental benefits of this manufacturing method?
A: The process emphasizes green chemistry principles by enabling solvent recycling. Solvents like ethanol, methanol, and ethyl acetate used in extraction and chromatography can be recovered and reused. This significantly reduces waste generation and operational costs associated with solvent disposal, aligning with modern environmental compliance standards.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Orlistat Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a supply chain that delivers both exceptional quality and economic efficiency. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory optimization to full-scale manufacturing is seamless. We are committed to maintaining stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of Orlistat meets the >99.5% purity threshold required for global regulatory submission. Our infrastructure is designed to support the complex crystallization and hydrogenation steps described in advanced patents, ensuring consistent product performance.
We invite you to collaborate with us to leverage these technological advancements for your product pipeline. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can enhance your supply chain security and reduce overall production costs. Let us be your partner in delivering high-quality anti-obesity therapeutics to the global market.