Advanced Solvent-Free Synthesis of Edaravone: Technical Breakthroughs and Commercial Scalability
Advanced Solvent-Free Synthesis of Edaravone: Technical Breakthroughs and Commercial Scalability
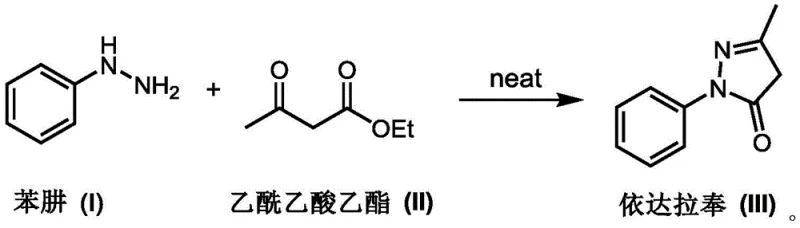
The pharmaceutical industry continuously seeks robust manufacturing processes that balance high purity with economic efficiency, particularly for critical neuroprotective agents like Edaravone. Patent CN113683566A introduces a transformative preparation method that fundamentally alters the synthesis landscape by eliminating the need for initial reaction solvents. This innovation addresses long-standing challenges in the production of 3-methyl-1-phenyl-2-pyrazolin-5-one, offering a pathway to achieve exceptional purity levels exceeding 99% while drastically simplifying the operational workflow. By shifting from traditional solvent-heavy protocols to a streamlined solvent-free cyclization followed by controlled crystallization, this technology represents a significant leap forward in green chemistry and process intensification for fine chemical manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of Edaravone has been plagued by inefficiencies inherent to solvent-dependent cyclization reactions. Conventional techniques typically involve refluxing phenylhydrazine and ethyl acetoacetate in high-boiling organic solvents or acidic aqueous media, which necessitates rigorous energy input to maintain elevated temperatures over extended periods. A critical drawback of these legacy methods is the formation of deeply colored crude products, mandating the use of activated carbon for decolorization and multiple recrystallization steps to meet pharmacopeial standards. These additional purification stages not only result in substantial product loss, thereby depressing overall yield, but also generate significant volumes of hazardous waste that complicate environmental compliance and increase disposal costs for manufacturers.
The Novel Approach
In stark contrast, the methodology disclosed in CN113683566A leverages a solvent-free environment to drive the condensation reaction, fundamentally changing the thermodynamic and kinetic profile of the synthesis. By mixing the reactants directly and heating the neat mixture to a moderate 75-80°C, the process achieves complete conversion within a concise 4 to 8-hour window without the thermal stress associated with high-boiling solvents. The resulting crude product emerges as a white solid powder, effectively bypassing the need for activated carbon treatment and complex refining sequences. This direct route not only preserves the structural integrity of the molecule but also ensures that the final isolated material possesses a purity greater than 99%, ready for downstream formulation with minimal intervention.
Mechanistic Insights into Solvent-Free Cyclization
The core of this technological advancement lies in the precise control of stoichiometry and thermal conditions during the solvent-free phase. The reaction proceeds through a nucleophilic attack of the hydrazine nitrogen on the carbonyl carbon of the beta-keto ester, followed by intramolecular cyclization and elimination of ethanol. Operating without a bulk solvent increases the effective concentration of the reactants, which accelerates the reaction rate according to the principles of collision theory. Furthermore, maintaining the temperature at an optimized 75°C prevents the thermal degradation of sensitive intermediates that often occurs at higher temperatures required for solvent reflux, thereby minimizing the formation of polymeric byproducts and tarry residues that typically contribute to product discoloration.
Impurity control is inherently managed through the selection of the crystallization solvent in the second stage. By introducing specific ethers or esters, such as methyl tert-butyl ether, post-reaction, the solubility profile of the target molecule is manipulated to favor the precipitation of pure Edaravone while keeping soluble impurities in the mother liquor. This selective crystallization mechanism is crucial for achieving the reported purity levels of over 99.4% directly from the filter cake. The absence of water during the initial cyclization step also mitigates hydrolysis side reactions, ensuring that the ester functionality is preserved until the intended cyclization event occurs, leading to a cleaner reaction profile and a more consistent impurity spectrum suitable for regulatory filing.
How to Synthesize Edaravone Efficiently
Implementing this synthesis route requires strict adherence to the specified molar ratios and thermal profiles to maximize the benefits of the solvent-free protocol. The process begins with the vigorous mixing of phenylhydrazine and ethyl acetoacetate at ambient temperatures to ensure homogeneity before the application of heat. Once the exothermic mixing phase is controlled, the system is heated to the target range to drive the cyclization to completion. Following the reaction, the strategic addition of a crystallization solvent allows for the isolation of the product in high yield. For detailed operational parameters and specific equipment configurations required for this synthesis, please refer to the standardized guide below.
- Mix phenylhydrazine and ethyl acetoacetate at a molar ratio of 1: 1.3 and stir vigorously at 0-30°C.
- Heat the mixture to 75-80°C and maintain for 4-8 hours under solvent-free conditions to ensure complete conversion.
- Add a crystallization solvent such as methyl tert-butyl ether, cool to 23-28°C, and filter to isolate white solid Edaravone with >99% purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this solvent-free technology translates into tangible operational efficiencies and risk mitigation strategies. The elimination of bulk solvents during the primary reaction phase significantly reduces the raw material inventory requirements and lowers the logistical burden associated with solvent transport and storage. Furthermore, the simplified workup procedure, which removes the need for activated carbon filtration and multiple recrystallizations, shortens the overall production cycle time. This acceleration in throughput enhances manufacturing capacity without the need for capital expenditure on new reactors, allowing suppliers to respond more agilely to market demand fluctuations for this critical neurological therapeutic intermediate.
- Cost Reduction in Manufacturing: The economic impact of this process is driven primarily by the drastic reduction in solvent consumption and waste treatment costs. By avoiding the use of large volumes of high-boiling solvents and eliminating the activated carbon decolorization step, manufacturers can achieve substantial savings in both raw material procurement and hazardous waste disposal fees. Additionally, the higher yield obtained through the minimization of purification losses directly improves the cost-of-goods-sold (COGS), making the final API more competitive in the global marketplace while maintaining healthy profit margins for producers.
- Enhanced Supply Chain Reliability: Supply continuity is bolstered by the robustness of the solvent-free method, which is less susceptible to variations in solvent quality or availability. The reliance on readily available starting materials like phenylhydrazine and ethyl acetoacetate, combined with a process that tolerates minor fluctuations without compromising purity, ensures a stable production output. This reliability is critical for downstream pharmaceutical customers who require consistent quality and uninterrupted supply to maintain their own finished drug product inventories and meet patient needs globally.
- Scalability and Environmental Compliance: From an environmental perspective, the reduction in solvent usage aligns perfectly with increasingly stringent global regulations regarding volatile organic compound (VOC) emissions. The process generates significantly less liquid waste, simplifying the effluent treatment requirements and reducing the environmental footprint of the manufacturing site. This green chemistry advantage not only facilitates easier regulatory approval but also future-proofs the supply chain against tightening environmental laws, ensuring long-term operational viability and sustainability for the manufacturing partner.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel Edaravone synthesis method. These insights are derived directly from the experimental data and process descriptions found in the patent literature, providing a clear understanding of the operational realities and benefits. Understanding these details is essential for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the value proposition of this manufacturing route.
Q: How does the solvent-free method improve Edaravone purity compared to traditional methods?
A: Traditional methods often require high-boiling solvents and activated carbon decolorization, which can introduce impurities and reduce yield. The patented solvent-free approach operates at moderate temperatures (75°C) and yields a white solid directly, eliminating the need for decolorization and achieving purity levels exceeding 99% without complex recrystallization.
Q: What are the specific reaction conditions for optimal yield in this process?
A: Optimal results are achieved by maintaining a molar ratio of phenylhydrazine to ethyl acetoacetate at 1:1.3. The reaction is initiated at room temperature and then heated to 75°C for approximately 6 hours. Subsequent crystallization using solvents like methyl tert-butyl ether at 26°C ensures high recovery rates.
Q: Is this synthesis method scalable for industrial production?
A: Yes, the process is highly scalable. By eliminating the need for large volumes of reaction solvents during the cyclization step, the method reduces reactor volume requirements and waste generation (E-factor). The simplified workup involving direct crystallization facilitates easier scale-up from laboratory to commercial tonnage.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Edaravone Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient and high-quality intermediate synthesis in the modern pharmaceutical supply chain. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative processes like the solvent-free Edaravone synthesis can be seamlessly transitioned from the laboratory to full-scale manufacturing. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs that utilize advanced analytical techniques to verify every batch, guaranteeing that the material you receive meets the highest international standards for safety and efficacy.
We invite you to collaborate with us to leverage this advanced technology for your supply chain needs. Our technical team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this optimized route can enhance your bottom line. Please contact our technical procurement team today to request specific COA data and comprehensive route feasibility assessments, and let us help you secure a sustainable and cost-effective supply of high-purity Edaravone for your global operations.