Advanced Manufacturing of Vildagliptin: A Technical Breakthrough for Commercial Scale-up
Introduction to Patent CN111138334A and Technological Breakthroughs
The pharmaceutical industry continuously seeks robust and scalable synthetic routes for critical antidiabetic agents, and the disclosure found in patent CN111138334A represents a significant advancement in the manufacturing of Vildagliptin. This specific intellectual property outlines a refined preparation method that addresses longstanding challenges associated with the synthesis of this selective dipeptide-peptidase-4 (DPP-4) inhibitor, which is crucial for managing type 2 diabetes by increasing endogenous incretin levels. The core innovation lies in a streamlined three-step sequence starting from L-prolinamide, moving through a chloroacetylation and dehydration phase, and culminating in a highly selective nucleophilic substitution with a functionalized adamantane derivative. By optimizing stoichiometric ratios and reaction parameters, this methodology offers a pathway to achieve superior chemical purity while mitigating the formation of persistent by-products that have historically plagued commercial production lines. For global procurement teams and R&D directors, understanding the nuances of this patent is essential for securing a reliable vildagliptin supplier capable of meeting stringent regulatory standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art techniques, such as those described in WO2011101861A1, often rely on reaction conditions that are energetically demanding and chemically inefficient, leading to suboptimal outcomes in large-scale manufacturing environments. In these traditional pathways, the final coupling reaction between the chloroacetyl pyrrolidine derivative and the adamantane amine frequently suffers from poor selectivity, resulting in a complex mixture of mono-substituted products and undesirable di-substituted impurities on the nitrogen atoms of the adamantane cage. The presence of these structurally similar impurities necessitates harsh control conditions and extensive purification protocols, often involving multiple recrystallization steps that drastically reduce the overall isolation yield to less than 30 percent in some reported instances. Furthermore, the synthetic routes for key intermediates in legacy methods are often elongated, requiring additional protection and deprotection steps that increase material costs, extend production lead times, and generate substantial volumes of chemical waste, thereby complicating cost reduction in API manufacturing efforts.
The Novel Approach
In stark contrast, the methodology presented in the subject patent introduces a direct and efficient strategy that eliminates the need for amino protecting groups, thereby fundamentally simplifying the molecular construction of the target compound. By carefully controlling the feeding ratio of L-prolinamide to chloroacetyl chloride at 1:3 and utilizing specific dehydration agents like TFAA, the process ensures high conversion rates while allowing for the easy removal of excess reagents through reduced pressure distillation. The subsequent nucleophilic substitution is conducted under moderate thermal conditions in tetrahydrofuran, which facilitates a clean reaction profile that inherently suppresses the formation of disubstituted side products. This strategic avoidance of protecting group chemistry not only shortens the synthetic timeline but also enhances the overall atom economy, making it an ideal candidate for the commercial scale-up of complex pharmaceutical intermediates where operational simplicity translates directly to margin improvement.
Mechanistic Insights into Acylation and Nucleophilic Substitution
The chemical transformation begins with the acylation of L-prolinamide, where the nucleophilic nitrogen of the pyrrolidine ring attacks the electrophilic carbonyl carbon of chloroacetyl chloride. The use of a three-fold excess of the acid chloride is a critical mechanistic driver that pushes the equilibrium towards the formation of (S)-N-chloroacetyl-2-carbamoyl pyrrolidine, ensuring that the starting amine is fully consumed to prevent downstream contamination. Following this, the dehydration step utilizes trifluoroacetic anhydride (TFAA) to convert the primary amide functionality into a nitrile group, a transformation that proceeds over an extended period of 8 hours to guarantee complete conversion without degrading the sensitive chloroacetyl moiety. This generates the highly reactive electrophile, (S)-N-chloroacetyl-2-cyano pyrrolidine, which is primed for the final coupling event.
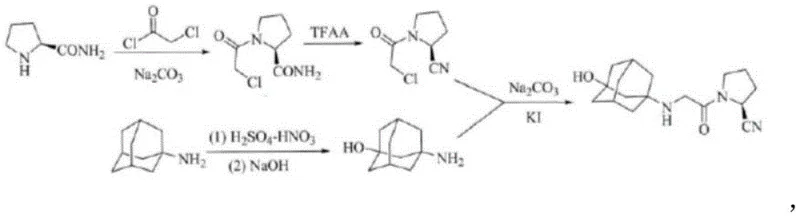
The final stage involves a nucleophilic substitution reaction where the primary amine of 3-amino-1-adamantanol attacks the methylene carbon adjacent to the chlorine atom in the pyrrolidine intermediate. The steric bulk of the adamantane cage usually poses a challenge for such substitutions; however, the specific solvent system and temperature control at 60°C provide the necessary activation energy to overcome this barrier while maintaining selectivity. Crucially, the absence of protecting groups on the adamantane amine prevents competitive dialkylation, a common side reaction in similar systems, thus ensuring that the nitrogen atom remains mono-substituted. The reaction mixture is then treated with methyl tert-butyl ether, which acts as an anti-solvent to induce rapid crystallization of the product within 10 minutes, effectively locking the high-purity crystal lattice and excluding soluble impurities from the final solid matrix.
How to Synthesize Vildagliptin Efficiently
Implementing this synthesis route requires precise adherence to the stoichiometric and temporal parameters defined in the patent to maximize yield and minimize impurity profiles. The process is designed to be operationally straightforward, leveraging standard unit operations such as distillation, heating, and precipitation that are universally available in modern fine chemical facilities. Operators must pay close attention to the removal of excess chloroacetyl chloride prior to the dehydration step to prevent side reactions, and strictly maintain the 60°C temperature window during the coupling phase to ensure optimal kinetics. For a detailed breakdown of the standardized operating procedures and safety protocols required for execution, please refer to the technical guide below.
- Acylate L-prolinamide with chloroacetyl chloride using a 1: 3 molar ratio to form (S)-N-chloroacetyl-2-carbamoyl pyrrolidine, removing excess reagent via distillation.
- Dehydrate the carbamoyl intermediate using TFAA over an 8-hour period to generate the key nitrile species, (S)-N-chloroacetyl-2-cyano pyrrolidine.
- Perform nucleophilic substitution with 3-amino-1-adamantanol in THF at 60°C for 6 hours, followed by precipitation with methyl tert-butyl ether to isolate the product.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, the adoption of this patented synthesis method offers profound benefits that extend beyond mere technical feasibility, directly impacting the bottom line and supply chain resilience for pharmaceutical manufacturers. By simplifying the synthetic route and eliminating complex purification steps, the process inherently reduces the consumption of solvents and reagents, leading to substantial cost savings in raw material procurement and waste disposal. The robustness of the reaction conditions allows for greater flexibility in production scheduling, reducing the risk of batch failures that can disrupt supply continuity and delay time-to-market for critical diabetes medications. Furthermore, the high selectivity of the reaction minimizes the need for expensive chromatographic purification media, which are often a bottleneck in the manufacturing of high-purity pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The elimination of protecting group strategies significantly lowers the number of synthetic steps, which directly correlates to reduced labor costs, lower energy consumption, and decreased solvent usage across the entire production lifecycle. By avoiding the generation of difficult-to-separate di-substituted impurities, the process reduces the loss of valuable material during purification, thereby improving the effective yield and lowering the cost per kilogram of the active ingredient. Additionally, the ability to remove excess reagents via simple distillation rather than complex aqueous workups further streamlines the operational expenditure, making this route economically superior to legacy methods that require extensive resource allocation for waste treatment and product recovery.
- Enhanced Supply Chain Reliability: The reliance on readily available starting materials such as L-prolinamide and chloroacetyl chloride ensures that the supply chain is not vulnerable to shortages of exotic or highly specialized reagents. The simplified process flow reduces the dependency on third-party vendors for custom intermediates, allowing manufacturers to exert greater control over their inventory levels and production timelines. Moreover, the rapid precipitation of the final product using methyl tert-butyl ether accelerates the isolation phase, shortening the overall cycle time per batch and enabling facilities to respond more agilely to fluctuations in market demand for Vildagliptin without compromising on quality or delivery commitments.
- Scalability and Environmental Compliance: The process is inherently designed for scalability, utilizing reaction conditions that are easily transferable from pilot plant to multi-ton commercial reactors without significant re-engineering. The reduction in by-product formation aligns with green chemistry principles by minimizing the environmental footprint associated with pharmaceutical manufacturing, specifically regarding the volume of hazardous waste generated. This compliance with environmental standards not only mitigates regulatory risks but also enhances the corporate sustainability profile of the manufacturer, which is increasingly becoming a key criterion for selection by major multinational pharmaceutical companies seeking responsible and eco-friendly partners for their API supply chains.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology, providing clarity for stakeholders evaluating its potential for integration into their existing manufacturing portfolios. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, ensuring that the information provided is accurate and relevant to real-world production scenarios. Understanding these details is vital for making informed decisions about technology transfer and process validation.
Q: How does this new method improve product purity compared to prior art?
A: The novel approach avoids the use of protecting groups on the amino group of the adamantane derivative, which significantly reduces the formation of difficult-to-remove disubstituted impurities on the nitrogen atom, thereby simplifying purification and enhancing final API quality.
Q: What are the critical reaction conditions for the final coupling step?
A: The final nucleophilic substitution is optimally conducted in tetrahydrofuran (THF) at a controlled temperature of 60°C for approximately 6 hours, utilizing methyl tert-butyl ether as an anti-solvent to induce rapid precipitation of the solid product within 10 minutes.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the process is designed for scalability with simple operational steps, readily available raw materials like L-prolinamide and chloroacetyl chloride, and robust workup procedures that eliminate the need for complex chromatographic separations typically required in older methods.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Vildagliptin Supplier
At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure necessary to translate the innovations described in patent CN111138334A into commercial reality, serving as a trusted partner for global pharmaceutical companies. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We maintain stringent purity specifications through our rigorous QC labs, employing advanced analytical techniques to monitor every critical quality attribute of the Vildagliptin intermediate and final API, guaranteeing that every batch meets the highest international standards for safety and efficacy.
We invite you to engage with our technical procurement team to discuss how this advanced synthesis route can be tailored to your specific volume requirements and cost targets. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits specific to your supply chain configuration. We encourage potential partners to contact us directly to obtain specific COA data and route feasibility assessments, allowing you to validate the superior quality and reliability of our manufacturing capabilities before making any long-term commitments.