Advanced Silylation Strategy for High-Purity Vildagliptin Manufacturing and Commercial Scale-Up
The pharmaceutical industry's relentless pursuit of efficient, scalable, and high-purity synthetic routes for dipeptidyl peptidase-4 (DPP-IV) inhibitors has led to significant innovations in process chemistry. Patent CN110642769B, published in March 2022, introduces a transformative preparation method for Vildagliptin, a critical antidiabetic agent. This technology addresses long-standing challenges in the alkylation of 3-aminoadamantanol, specifically targeting the suppression of the notorious bis-alkylated byproduct, Impurity Ia. By employing a strategic silylation protection protocol using 1,1,1,3,3,3-hexamethyldisilazane (HMDS), the process achieves a remarkable purity profile with Impurity Ia levels as low as 0.03%, alongside yields exceeding 85%. For global supply chain leaders and R&D directors, this represents a pivotal shift from laborious purification techniques to a streamlined, industrially viable workflow that enhances both economic efficiency and product quality.
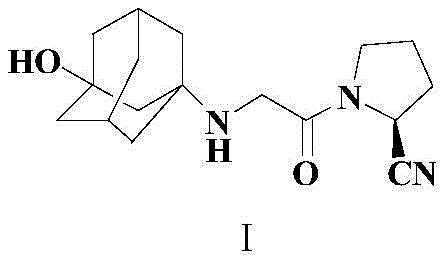
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for Vildagliptin, such as those described in WO0034241 and US2008167479, typically involve the direct nucleophilic substitution of 3-aminoadamantanol with an alpha-chloro ketone derivative, specifically (S)-1-(2-chloroacetyl)pyrrolidine-2-carbonitrile. While conceptually straightforward, this direct alkylation suffers from a fundamental chemoselectivity issue: the product itself contains a secondary amine that remains nucleophilic under the reaction conditions. Consequently, if even a slight excess of the alkylating agent is present, or if the reaction kinetics favor over-alkylation, the secondary amine reacts further to form the tertiary amine byproduct, known as Impurity Ia. This side reaction is thermodynamically favorable and difficult to suppress in a one-pot direct alkylation, often resulting in crude products containing greater than 0.1% of this structurally similar impurity.
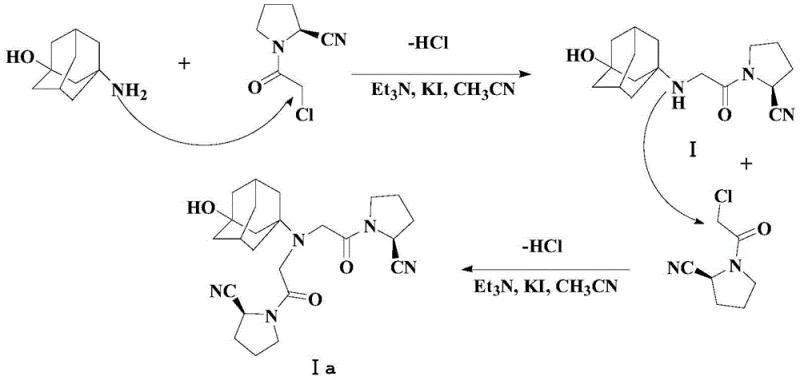
The presence of Impurity Ia poses severe downstream processing challenges. Because Impurity Ia shares significant structural homology with the target Vildagliptin, separating them requires rigorous and costly purification steps, often involving column chromatography or multiple recrystallizations. In a commercial manufacturing setting, column chromatography is economically prohibitive due to high solvent consumption, silica gel costs, and low throughput. Furthermore, the need for repeated purification cycles drastically reduces the overall yield and extends the production lead time, creating bottlenecks for reliable API intermediate suppliers aiming to meet large-scale demand. The inability to effectively control this specific impurity profile has historically been a major pain point for procurement managers seeking cost reduction in pharmaceutical intermediate manufacturing.
The Novel Approach
The methodology disclosed in CN110642769B offers an elegant solution by fundamentally altering the reactivity of the starting material through temporary protection. Instead of exposing the naked amine of 3-aminoadamantanol directly to the alkylating agent, the process first subjects the substrate to silylation using HMDS. This step concurrently protects both the amino and hydroxyl functionalities, generating a bis-silylated intermediate (Compound II). This protection strategy serves a dual purpose: it masks the nucleophilicity of the nitrogen atom to prevent uncontrolled poly-alkylation and modifies the steric environment around the reactive center. When this protected intermediate is subsequently reacted with the chloroacetyl pyrrolidine derivative in the presence of potassium iodide and an organic base, the reaction proceeds with high selectivity.
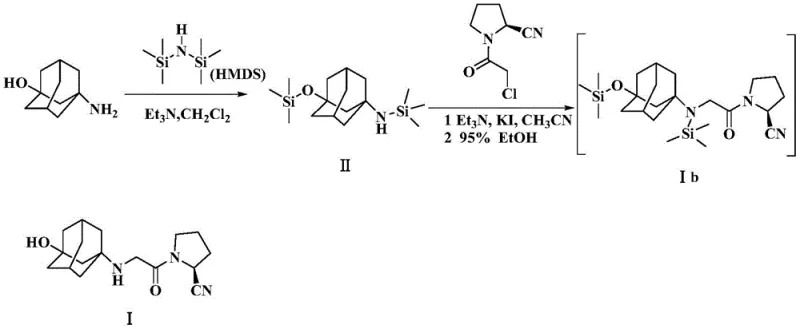
Crucially, the novel approach integrates the deprotection step into the workup phase, creating a highly efficient 'one-pot' sequence for the final transformation. After the alkylation is complete, the addition of 95% ethanol facilitates the rapid removal of the trimethylsilyl protecting groups under mild conditions. This cleverly timed deprotection ensures that the free secondary amine of Vildagliptin is only generated after the alkylating agent has been fully consumed or quenched, thereby kinetically trapping the reaction at the mono-alkylated stage and virtually eliminating the formation of Impurity Ia. This mechanistic refinement allows for the isolation of high-purity Vildagliptin (99.40% HPLC purity) through simple filtration and recrystallization, bypassing the need for chromatographic separation entirely and significantly simplifying the commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Silyl-Mediated Selective Alkylation
The core of this technological advancement lies in the modulation of nucleophilicity via silyl protection. In the conventional unprotected route, the pKa and steric profile of the 3-aminoadamantanol allow it to act as a potent nucleophile. However, once mono-alkylated, the resulting secondary amine retains sufficient nucleophilicity to attack a second equivalent of the electrophile. By converting the primary amine into a silylamine (N-Si bond) and the alcohol into a silyl ether (O-Si bond), the electron density on the nitrogen is significantly altered. While silylamines can still participate in nucleophilic substitutions, especially under catalysis by iodide ions which enhance the leaving group ability of the chloride via Finkelstein-type activation, the steric bulk of the silyl group imposes a kinetic barrier. This barrier is sufficient to allow the initial formation of the C-N bond but effectively shields the resulting nitrogen center from a second alkylation event while the silyl group remains attached.
Furthermore, the choice of reaction conditions plays a pivotal role in impurity control. The use of acetonitrile as a polar aprotic solvent facilitates the dissolution of the ionic intermediates and supports the SN2 mechanism required for the displacement of the chloride. The inclusion of potassium iodide acts as a phase-transfer-like catalyst or nucleophilic catalyst, generating a more reactive alkyl iodide species in situ, which lowers the activation energy for the desired substitution. Simultaneously, the organic base, such as triethylamine or diisopropylethylamine, serves to scavenge the hydrochloric acid generated during the reaction, preventing the protonation of the amine which would render it non-nucleophilic. The precise control of temperature (75-80°C) ensures that the reaction rate is optimized for the protected species without triggering thermal degradation or alternative decomposition pathways, resulting in a clean reaction profile that is robust enough for industrial replication.
How to Synthesize Vildagliptin Efficiently
The synthesis of Vildagliptin via this protected route is designed for operational simplicity and high throughput. The process begins with the silylation of 3-aminoadamantanol in dichloromethane, followed by solvent exchange and alkylation in acetonitrile. The final deprotection and crystallization are integrated into the workup, minimizing unit operations. This streamlined workflow is particularly advantageous for contract development and manufacturing organizations (CDMOs) looking to optimize their production lines for diabetes therapeutics. The detailed standardized synthesis steps, including specific molar ratios and temperature profiles, are outlined below to ensure reproducibility and quality compliance.
- Protect 3-aminoadamantanol using HMDS and a catalyst in dichloromethane at 35-40°C to form the silylated intermediate.
- React the protected intermediate with (S)-1-(2-chloroacetyl)pyrrolidine-2-carbonitrile in acetonitrile with KI and organic base at 75-80°C.
- Add 95% ethanol to the reaction mixture to remove the silyl protecting group, followed by filtration and recrystallization to isolate pure Vildagliptin.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this silylation-based methodology translates directly into tangible operational efficiencies and risk mitigation. The most significant advantage is the drastic simplification of the purification train. By eliminating the reliance on column chromatography, manufacturers can avoid the substantial costs associated with silica gel procurement, solvent recovery, and waste disposal. This shift to crystallization-based purification not only reduces the variable cost of goods sold (COGS) but also accelerates the batch cycle time, allowing for faster turnover and improved responsiveness to market demand fluctuations. The ability to produce high-purity material without complex chromatographic steps makes the supply chain more resilient and less prone to bottlenecks caused by purification capacity constraints.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven by the elimination of expensive purification media and the reduction in solvent usage intensity. Traditional methods requiring chromatography consume vast quantities of solvents which must be purchased, distilled, and disposed of, creating a heavy financial and environmental burden. By switching to a precipitation and recrystallization workflow, the process significantly lowers the E-factor (environmental factor), leading to substantial cost savings in waste management. Additionally, the high yield (over 85%) ensures better atom economy, meaning less raw material is wasted, further driving down the effective cost per kilogram of the active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions contributes to a more reliable supply of this critical API intermediate. The reagents used, such as HMDS and common organic bases, are commodity chemicals with stable global availability, reducing the risk of raw material shortages. Moreover, the tolerance of the process to slight variations in stoichiometry, thanks to the protective group strategy, ensures consistent batch-to-batch quality. This consistency is vital for maintaining regulatory compliance and avoiding costly batch rejections, thereby securing a steady flow of material for downstream drug formulation and ensuring reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: From a scale-up perspective, the exothermic nature of the reactions is manageable within standard stainless steel reactors, and the absence of chromatographic steps removes a major hurdle in transitioning from pilot plant to commercial tonnage production. The process aligns well with green chemistry principles by minimizing hazardous waste generation and solvent consumption. This environmental compatibility is increasingly important for meeting the stringent sustainability criteria set by multinational pharmaceutical companies. The simplified waste stream, primarily consisting of recyclable organic solvents and benign salts, facilitates easier regulatory approval and lowers the long-term liability associated with chemical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel Vildagliptin synthesis route. These insights are derived directly from the experimental data and process descriptions found in the patent literature, providing a clear understanding of the method's capabilities and limitations for potential partners and technical evaluators.
Q: How does the HMDS protection strategy reduce Impurity Ia in Vildagliptin synthesis?
A: The silylation of both the amino and hydroxyl groups on 3-aminoadamantanol prevents the secondary amine in the mono-alkylated product from reacting further with the chloroacetyl reagent, effectively blocking the formation of the bis-alkylated Impurity Ia.
Q: What are the critical reaction conditions for the alkylation step?
A: The alkylation requires heating to 75-80°C in acetonitrile with potassium iodide as a catalyst and an organic base like triethylamine to scavenge acid, ensuring complete conversion while maintaining selectivity.
Q: Is column chromatography required for purification in this new method?
A: No, the improved selectivity of this process allows for purification via simple filtration and recrystallization using solvents like ethanol and butanone, eliminating the need for expensive and time-consuming column chromatography.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Vildagliptin Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic technologies to maintain competitiveness in the global pharmaceutical market. Our team of expert chemists has thoroughly analyzed the silylation-based route for Vildagliptin and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We are committed to delivering high-purity intermediates that meet the most stringent purity specifications, utilizing our rigorous QC labs to ensure every batch complies with international pharmacopoeia standards. Our facility is equipped to handle the specific solvent systems and reaction conditions required for this optimized process, ensuring a seamless transition from laboratory innovation to industrial reality.
We invite procurement directors and R&D leaders to collaborate with us to leverage this cost-effective and high-yielding technology for your supply chain. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, ensuring that your project benefits from the highest standards of quality and efficiency in the industry.