Scalable Synthesis of Voglibose Impurity I: Advanced Process Control for Global Quality Standards
Scalable Synthesis of Voglibose Impurity I: Advanced Process Control for Global Quality Standards
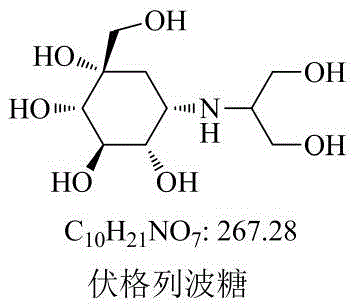
The pharmaceutical landscape for diabetes management relies heavily on alpha-glucosidase inhibitors, with Voglibose standing as a cornerstone therapy for controlling postprandial hyperglycemia. As regulatory bodies worldwide, including those enforcing the Japanese Pharmacopoeia (JP17) and Chinese Pharmacopoeia, tighten their specifications for drug purity, the availability of certified reference standards for related impurities becomes a critical bottleneck in quality assurance. Patent CN110511152B discloses a robust and novel preparation method for Voglibose Impurity I hydrochloride, addressing a significant gap in the existing technical literature where specific synthetic routes for this isomer were previously undefined. This technological breakthrough not only facilitates accurate qualitative and quantitative analysis but also empowers manufacturers to establish stricter internal quality controls, thereby enhancing the overall medication safety profile of Voglibose formulations in the global market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
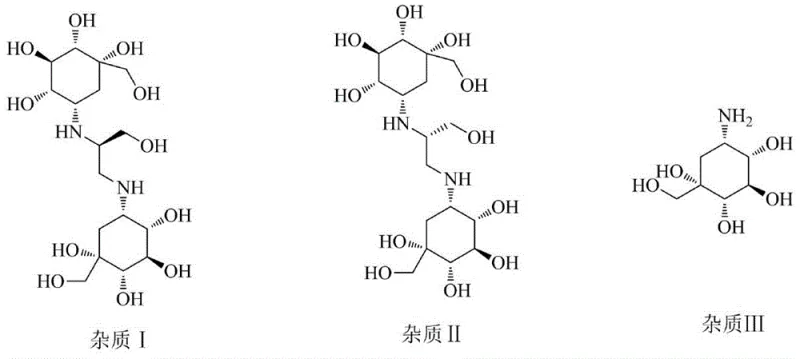
Prior to the disclosure of this specific methodology, the industry faced substantial challenges in sourcing authentic samples of Voglibose Impurity I. While the structures of potential impurities were documented in regulatory files, the lack of a defined synthetic pathway meant that quality control laboratories often struggled to differentiate between isomeric impurities, particularly Impurity I and Impurity II, which share identical molecular weights but differ in stereochemical configuration. Conventional approaches often relied on isolation from crude reaction mixtures, a process that is inherently inefficient, yields negligible quantities, and fails to provide the high purity required for analytical validation. Furthermore, without a dedicated synthesis route, the risk of misidentifying impurities during stability studies or batch release testing was elevated, potentially leading to regulatory non-compliance and costly product recalls.
The Novel Approach
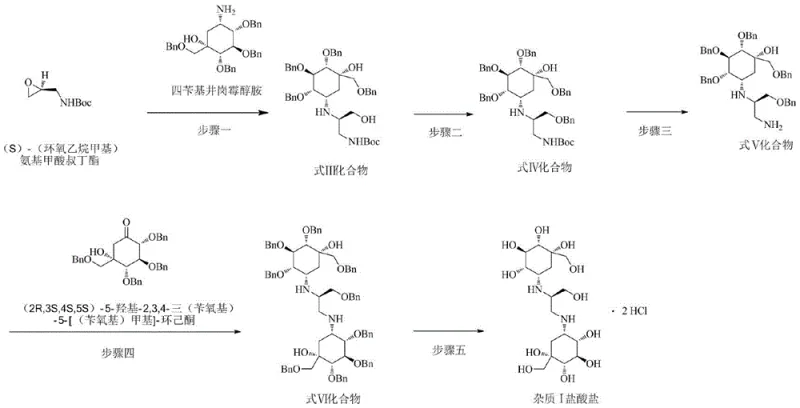
The methodology outlined in CN110511152B introduces a streamlined, five-step synthetic strategy that decisively overcomes these historical limitations by utilizing stereoselective starting materials to construct the specific isomer. By employing (S)-(oxiranylmethyl) carbamic acid tert-butyl ester as a chiral building block, the process ensures the correct spatial arrangement of the side chain, effectively distinguishing the target molecule from its isomers. This approach eliminates the need for difficult chromatographic separations of isomers at the final stage, as the stereochemistry is locked in early in the synthesis. The route is designed for scalability, utilizing standard organic transformations such as nucleophilic ring opening, protective group manipulation, and reductive amination, making it highly suitable for the commercial production of reference standards needed by reliable voglibose impurity supplier networks globally.
Mechanistic Insights into Stereoselective Reductive Amination
The core of this synthesis lies in the precise orchestration of protecting group chemistry and stereoselective bond formation. The process initiates with the nucleophilic attack of tetrabenzylvaliolamine on the less hindered carbon of the chiral epoxide ring. This ring-opening reaction is critical as it installs the nitrogen-containing side chain with the requisite (S)-configuration. Following this, the strategic use of benzyl protecting groups serves a dual purpose: it prevents unwanted side reactions at the hydroxyl moieties during subsequent steps and enhances the solubility of the intermediates in organic solvents, facilitating easier purification. The deprotection of the Boc group under acidic conditions reveals the primary amine, which is then poised for the pivotal coupling reaction with the cyclohexanone derivative.
The final coupling step utilizes sodium cyanoborohydride for reductive amination, a choice driven by its chemoselectivity. Unlike stronger reducing agents, sodium cyanoborohydride selectively reduces the iminium ion intermediate formed between the amine and the ketone without affecting other sensitive functional groups or causing over-reduction. This mechanistic precision is vital for maintaining the integrity of the complex polyol structure. The final catalytic hydrogenation step simultaneously removes all benzyl protecting groups and forms the hydrochloride salt. This telescoped deprotection-salification strategy is highly efficient, minimizing unit operations and reducing the potential for product degradation, thus ensuring the final material meets the stringent purity specifications demanded by modern analytical laboratories.
How to Synthesize Voglibose Impurity I Efficiently
The synthesis of this critical reference standard follows a logical progression of functional group transformations designed to maximize yield and stereochemical fidelity. The process begins with the coupling of the valiolamine core with the chiral epoxide, followed by protection, deprotection, and final assembly of the dimeric structure. Each step has been optimized to balance reaction kinetics with ease of workup, ensuring that the process is robust enough for technology transfer. For detailed operational parameters, stoichiometry, and specific workup procedures required to replicate this synthesis in a GMP environment, please refer to the standardized protocol below.
- Perform epoxide amination using (S)-(oxiranylmethyl) carbamic acid tert-butyl ester and tetrabenzylvaliolamine to obtain Compound III.
- Execute benzyl protection on the hydroxyl group of Compound III using benzyl chloride under basic conditions to yield Compound IV.
- Deprotect the amino group of Compound IV using acid (HCl or TFA) to generate the free amine Compound V.
- Conduct condensation reduction with a specific cyclohexanone derivative and sodium cyanoborohydride to form Compound VI.
- Finalize the process via catalytic hydrogenation deprotection and salification with hydrogen chloride gas to obtain the target hydrochloride salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers tangible strategic benefits beyond mere technical feasibility. The primary advantage lies in the simplification of the supply chain for critical quality control materials. By establishing a reliable in-house or contract manufacturing route for Impurity I, pharmaceutical companies reduce their dependency on scarce external vendors who may charge premium prices for small quantities of reference standards. The use of commodity chemicals such as benzyl chloride, sodium hydroxide, and palladium on carbon ensures that raw material availability is not a bottleneck, contributing to cost reduction in pharmaceutical intermediate manufacturing by stabilizing input costs and mitigating supply risks associated with exotic reagents.
- Cost Reduction in Manufacturing: The synthetic route avoids the use of expensive transition metal catalysts (other than standard Pd/C) or cryogenic conditions, which significantly lowers the operational expenditure (OPEX) associated with production. The high molar yields reported in the patent examples, particularly in the deprotection and final salification steps, indicate a material-efficient process that minimizes waste generation. By eliminating the need for complex chiral separations at the end of the synthesis, the process reduces solvent consumption and chromatography resin costs, leading to substantial overall cost savings compared to isolation-based methods.
- Enhanced Supply Chain Reliability: The reliance on stable, shelf-stable intermediates and widely available reagents ensures that production schedules are not disrupted by raw material shortages. The robustness of the benzyl protection strategy allows intermediates to be stored and transported with minimal degradation risk, providing flexibility in production planning. This reliability is crucial for maintaining continuous quality control operations, ensuring that reducing lead time for high-purity voglibose impurities becomes a reality rather than just a goal, thereby supporting faster batch release cycles for the final drug product.
- Scalability and Environmental Compliance: The process is inherently scalable, moving seamlessly from gram-scale laboratory synthesis to multi-kilogram production without fundamental changes in reaction chemistry. The use of catalytic hydrogenation for the final deprotection is a green chemistry principle in action, generating water and toluene/cyclohexane as byproducts rather than stoichiometric chemical waste. This aligns with modern environmental, social, and governance (ESG) goals, simplifying waste treatment protocols and reducing the environmental footprint of the manufacturing process, which is increasingly important for commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Voglibose Impurity I. These insights are derived directly from the patent specifications and are intended to clarify the value proposition of this synthesis method for stakeholders involved in quality assurance and process development. Understanding these nuances is essential for integrating this reference standard into your broader analytical control strategy.
Q: Why is the preparation of Voglibose Impurity I critical for regulatory compliance?
A: Pharmacopoeias such as JP17 require strict control of related substances. Since Impurity I is a known isomer that can co-elute or interfere with analysis, having an authentic reference standard is mandatory for validating HPLC methods and ensuring batch release compliance.
Q: What represents the key stereochemical challenge in this synthesis?
A: The synthesis relies on the precise stereochemistry of the starting epoxide, specifically (S)-(oxiranylmethyl) carbamate. Maintaining this configuration throughout the amination and subsequent coupling steps is essential to distinguish Impurity I from its isomer, Impurity II.
Q: How does this method improve supply chain stability for reference standards?
A: By utilizing commercially available starting materials like tetrabenzylvaliolamine and avoiding exotic catalysts, the route ensures consistent reproducibility. The robust five-step sequence minimizes purification bottlenecks, allowing for reliable production of high-purity reference materials.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Voglibose Impurity I Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your final drug product depends on the quality of every component, including the reference standards used to test it. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that whether you need grams for method validation or kilograms for stability programs, we can deliver. Our rigorous QC labs and commitment to stringent purity specifications mean that every batch of Voglibose Impurity I we supply is accompanied by comprehensive analytical data, giving you the confidence to meet the toughest regulatory audits.
We invite you to leverage our technical expertise to optimize your supply chain for critical impurities. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our advanced synthesis capabilities can support your long-term quality and cost objectives.