Scalable Manufacturing of Tapentadol Hydrochloride Intermediates via Novel Deprotection
Scalable Manufacturing of Tapentadol Hydrochloride Intermediates via Novel Deprotection
The global demand for central nervous system analgesics continues to drive innovation in the synthesis of opioid receptor agonists, particularly Tapentadol Hydrochloride. A pivotal advancement in this domain is detailed in patent CN103159633A, which discloses a highly efficient preparation method for Tapentadol Hydrochloride and its key intermediates. This technology addresses critical bottlenecks in traditional manufacturing by introducing a novel synthetic pathway that bypasses the need for expensive chiral column separation and avoids the use of highly corrosive demethylation reagents. By leveraging an allyl protection strategy combined with catalytic hydrogenolysis, the process achieves superior stereochemical control and operational safety. For procurement leaders and R&D directors seeking a reliable pharmaceutical intermediates supplier, understanding the mechanistic advantages of this route is essential for securing a stable and cost-effective supply chain.
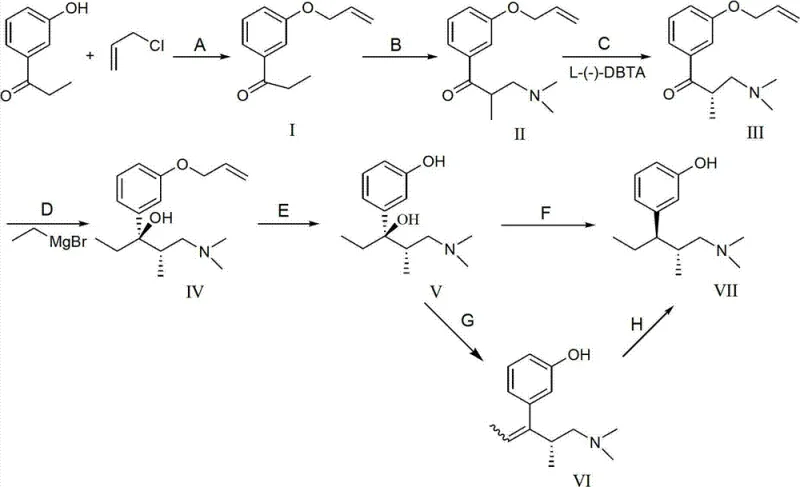
The significance of this patent lies in its ability to streamline the production of high-purity active pharmaceutical ingredients (APIs). Traditional routes often suffer from low yields and environmental hazards due to the use of volatile acids and heavy metal catalysts. In contrast, the methodology outlined in CN103159633A utilizes readily available starting materials such as 3-hydroxypropiophenone and employs standard unit operations like crystallization and filtration. This shift not only enhances the purity profile of the final product but also aligns with modern green chemistry principles, making it a compelling option for cost reduction in pharmaceutical intermediates manufacturing. The following analysis dissects the technical nuances and commercial implications of this breakthrough synthesis.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historical synthetic routes for Tapentadol, such as those described in patent EP0693475, have long been plagued by significant operational inefficiencies and safety concerns. These conventional methods typically rely on a methyl group as a phenolic protecting group, which necessitates the use of concentrated hydrobromic acid or hydrochloric acid for the final demethylation step. This requirement introduces severe challenges, including the corrosion of reactor equipment, the generation of hazardous waste streams, and the difficulty in handling volatile acids on a large scale. Furthermore, achieving the necessary stereochemical purity in these older routes often mandates chiral column chromatography, a technique that is notoriously expensive, difficult to scale, and prone to product loss. The combination of harsh reaction conditions and complex purification steps results in a fragmented supply chain with elevated production costs and inconsistent batch-to-batch quality.
The Novel Approach
The innovative strategy presented in CN103159633A fundamentally reengineers the synthesis by substituting the methyl protecting group with an allyl group. This seemingly simple modification unlocks a cascade of process improvements, primarily centered around the deprotection step. Instead of aggressive acid hydrolysis, the allyl group is removed via catalytic hydrogenolysis under mild conditions, typically using palladium on carbon (Pd/C) and ammonium formate or hydrogen gas. This approach eliminates the need for corrosive acids entirely, thereby extending equipment lifespan and simplifying waste management. Additionally, the route incorporates a chiral resolution step using L-(-)-dibenzoyl tartaric acid (L-(-)-DBTA) prior to the Grignard reaction, which allows for the isolation of the desired enantiomer through crystallization rather than chromatography. This transition from chromatographic separation to crystallization-based resolution is a game-changer for commercial scale-up of complex pharmaceutical intermediates, offering a robust and economically viable pathway to high-purity Tapentadol precursors.
Mechanistic Insights into Catalytic Hydrogenolysis and Chiral Resolution
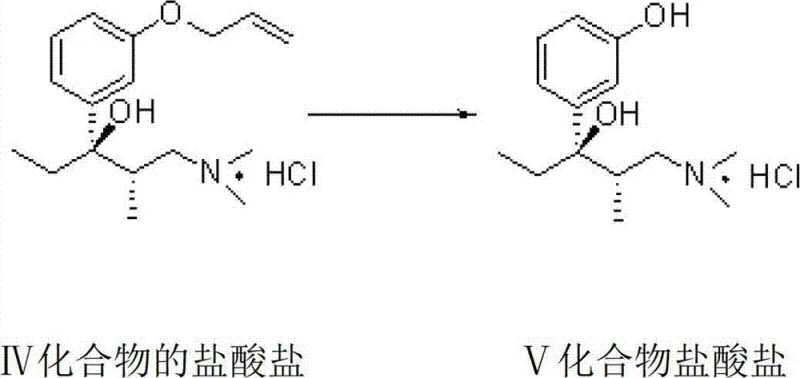
The core of this synthetic success lies in the precise orchestration of stereoselective reactions and protective group chemistry. The process begins with the O-alkylation of 3-hydroxypropiophenone using allyl chloride in the presence of a base and a catalyst like sodium iodide, forming the protected ketone intermediate. Following a Mannich reaction to introduce the dimethylamine moiety, the racemic mixture undergoes optical resolution. The use of L-(-)-dibenzoyl tartaric acid forms a diastereomeric salt with the (S)-enantiomer, which precipitates out of solution due to differences in solubility. This solid-state separation is far more efficient than liquid chromatography and sets the absolute configuration for the subsequent carbon-carbon bond-forming steps. The resolved intermediate then reacts with ethyl magnesium bromide in a Grignard addition, constructing the critical quaternary carbon center with high fidelity. The final transformation involves the removal of the allyl protecting group, where the pi-allyl palladium complex facilitates the cleavage of the C-O bond, regenerating the phenolic hydroxyl group without affecting the sensitive amine functionality or the newly formed stereocenters.
Impurity control is rigorously maintained throughout this sequence, particularly during the dehydration and hydrogenation phases that follow the formation of the key alcohol intermediate. The patent describes two divergent paths from the deprotected alcohol: one involving acid-catalyzed dehydration to an alkene followed by hydrogenation, and another involving acylation followed by hydrogenolysis. Both pathways are designed to minimize the formation of regioisomers and over-reduced byproducts. The mild conditions of the hydrogenolysis step ensure that the dimethylamine group remains intact, preventing N-demethylation which is a common side reaction in harsher acidic environments. This selectivity is crucial for meeting the stringent purity specifications required for high-purity pharmaceutical intermediates. By controlling parameters such as temperature (50-100°C for deprotection) and catalyst loading, the process ensures that impurity profiles remain well within acceptable limits, reducing the burden on downstream purification units.
How to Synthesize Tapentadol Hydrochloride Efficiently
The synthesis of Tapentadol Hydrochloride intermediates via this patented route involves a sequence of well-defined chemical transformations that prioritize yield and stereochemical integrity. The process initiates with the protection of the phenolic hydroxyl group, followed by amine introduction and chiral resolution to establish the correct configuration. Subsequent Grignard addition builds the carbon skeleton, and the critical deprotection step unveils the active phenol. For a detailed breakdown of the specific reaction conditions, stoichiometry, and workup procedures required to execute this synthesis in a GMP environment, please refer to the standardized protocol below.
- Perform allyl protection on 3-hydroxypropiophenone followed by a Mannich reaction to introduce the amine functionality.
- Execute chiral resolution using L-(-)-dibenzoyl tartaric acid to isolate the desired (S)-enantiomer intermediate.
- Conduct Grignard addition with ethyl magnesium bromide, followed by catalytic hydrogenolysis to remove the allyl protecting group under mild conditions.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of the synthesis route described in CN103159633A offers tangible strategic benefits that extend beyond mere technical feasibility. The primary advantage is the substantial reduction in manufacturing costs driven by the elimination of chiral chromatography. Chromatographic separations are capital-intensive and consume significant amounts of solvent and stationary phase; replacing this with crystallization drastically lowers the cost of goods sold (COGS). Furthermore, the removal of corrosive demethylation reagents like concentrated hydrobromic acid reduces the need for specialized Hastelloy reactors and minimizes the costs associated with hazardous waste disposal. These factors combine to create a more resilient and cost-efficient supply chain, ensuring that reducing lead time for high-purity pharmaceutical intermediates becomes a achievable reality rather than just a goal.
- Cost Reduction in Manufacturing: The substitution of methyl protection with allyl protection allows for the use of catalytic hydrogenation instead of stoichiometric acid demethylation. This change eliminates the consumption of expensive and hazardous acids, reduces reactor maintenance costs due to corrosion, and simplifies the neutralization and waste treatment processes. Additionally, the high yields reported in the patent examples for each step contribute to a more efficient use of raw materials, further driving down the overall production cost per kilogram of the final API.
- Enhanced Supply Chain Reliability: By relying on widely available starting materials such as 3-hydroxypropiophenone and allyl chloride, the process mitigates the risk of raw material shortages. The robustness of the crystallization-based resolution step ensures consistent supply of the correct enantiomer without dependence on scarce chiral columns or specialized separation services. This stability is critical for maintaining continuous production schedules and meeting the rigorous delivery timelines demanded by global pharmaceutical clients.
- Scalability and Environmental Compliance: The process is inherently scalable, utilizing common organic solvents like acetone, ethanol, and ethyl acetate which are easily recovered and recycled. The avoidance of heavy metal contaminants from harsh acid treatments and the reduction of hazardous waste streams align with increasingly strict environmental regulations. This eco-friendly profile not only facilitates regulatory approval but also enhances the corporate sustainability metrics of the manufacturing partner, making it a preferred choice for environmentally conscious buyers.
Frequently Asked Questions (FAQ)
The following questions address common inquiries regarding the technical implementation and commercial viability of this synthesis route. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on how this method compares to existing technologies and what specific advantages it offers for industrial application.
Q: How does this patent improve upon traditional Tapentadol synthesis routes?
A: Unlike conventional methods that require harsh demethylation with concentrated hydrobromic acid and expensive chiral column separation, this process utilizes an allyl protecting group removable via mild catalytic hydrogenation and achieves chirality through crystallization-based resolution, significantly reducing equipment corrosion and operational costs.
Q: What represents the key cost-saving advantage in this manufacturing process?
A: The elimination of chiral column chromatography and the replacement of corrosive demethylation reagents with catalytic hydrogenolysis drastically lower both raw material costs and waste treatment expenses, while improving overall yield stability.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the process is specifically designed for industrialization, utilizing common solvents like acetone and ethanol, avoiding hazardous reagents, and demonstrating high yields in pilot examples, making it ideal for commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tapentadol Hydrochloride Supplier
The technological advancements detailed in patent CN103159633A represent a significant leap forward in the efficient production of Tapentadol Hydrochloride intermediates. At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure to translate these laboratory-scale innovations into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the benefits of this novel route—such as improved purity and reduced environmental impact—are fully realized at an industrial level. We operate stringent purity specifications and maintain rigorous QC labs to guarantee that every batch meets the highest international standards for pharmaceutical intermediates.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific supply chain needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic advantages of switching to this allyl-protection strategy. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project requirements, ensuring a seamless transition to a more efficient and sustainable manufacturing process.