Advanced Synthetic Route for Tapentadol Hydrochloride: Enhancing Purity and Commercial Scalability
Advanced Synthetic Route for Tapentadol Hydrochloride: Enhancing Purity and Commercial Scalability
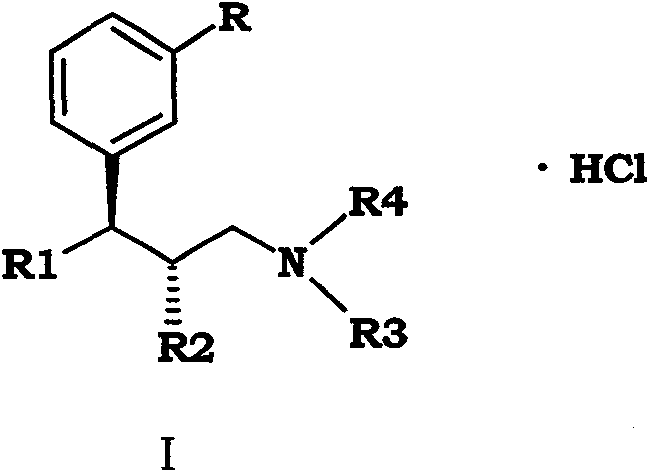
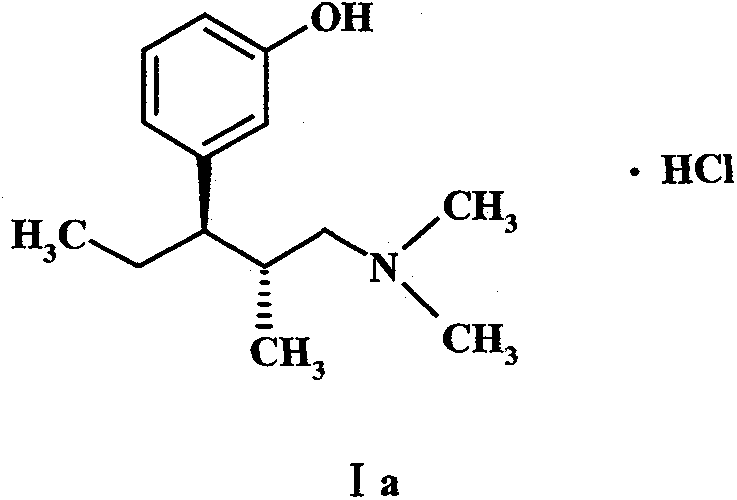
The pharmaceutical industry continuously seeks robust manufacturing pathways for potent analgesics like Tapentadol Hydrochloride, a dual-action mu-opioid receptor agonist and norepinephrine reuptake inhibitor. Patent CN102557851B introduces a transformative synthetic methodology that addresses critical bottlenecks in existing production technologies, specifically targeting the inefficiencies associated with traditional Grignard-based approaches. This innovation utilizes 1-(3-substituted-phenyl)-1-ketone compounds as accessible starting materials, facilitating a streamlined sequence that enhances overall yield while maintaining stringent purity standards required for active pharmaceutical ingredients. By shifting away from moisture-sensitive organometallic reagents, this route offers a safer, more controllable environment for chemical transformation, directly impacting the reliability of the supply chain for global healthcare providers. The strategic design of this synthesis ensures that intermediate isolation steps are minimized, thereby reducing solvent consumption and waste generation in alignment with modern green chemistry principles. For stakeholders evaluating long-term procurement strategies, understanding the mechanistic superiority of this patent is essential for securing a competitive edge in the analgesic market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of Tapentadol Hydrochloride has relied heavily on pathways involving Grignard reactions and high-pressure catalytic hydrogenation, as illustrated in various prior art documents such as WO2008/012047 and WO2011/067714A1. These conventional methods impose severe operational constraints, requiring strictly anhydrous conditions and specialized high-pressure equipment that significantly escalate capital expenditure and maintenance costs for manufacturing facilities. Furthermore, the use of hazardous reagents like borane dimethyl sulfide or expensive acylating agents introduces substantial safety risks and environmental compliance challenges, complicating the waste treatment processes necessary for regulatory approval. The reliance on chiral liquid chromatography for separation in some older routes further exacerbates production costs, rendering them economically unviable for large-scale commercialization without significant process optimization. Consequently, supply chain continuity is often threatened by the complexity of these operations, leading to potential delays and inconsistent batch quality that can disrupt downstream formulation schedules for pharmaceutical companies. These cumulative drawbacks highlight the urgent need for a more resilient and cost-effective synthetic strategy that can withstand the rigors of modern industrial production.
The Novel Approach
In stark contrast to legacy technologies, the method disclosed in CN102557851B employs a nucleophilic substitution strategy using beta-dicarbonyl compounds, effectively bypassing the need for dangerous Grignard reagents and high-pressure hydrogenation steps. This innovative route leverages readily available raw materials, such as 1-(3-methoxy-phenyl)-1-propanone, which are commercially abundant and cost-stable, ensuring a reliable feedstock supply for continuous manufacturing operations. The reaction conditions are notably mild, typically operating within a temperature range of 0°C to 150°C depending on the specific step, which reduces energy consumption and minimizes the formation of thermal degradation byproducts. By integrating a chiral resolution step using L-(-)-dibenzoyl tartaric acid rather than complex chromatographic separation, the process achieves high enantiomeric purity with significantly lower operational overhead. This structural simplification of the synthetic pathway not only enhances safety profiles for plant operators but also facilitates easier technology transfer between different manufacturing sites globally. Ultimately, this approach represents a paradigm shift towards sustainable and scalable pharmaceutical manufacturing, aligning perfectly with the strategic goals of cost reduction in API manufacturing.
Mechanistic Insights into Malonate Alkylation and Chiral Resolution
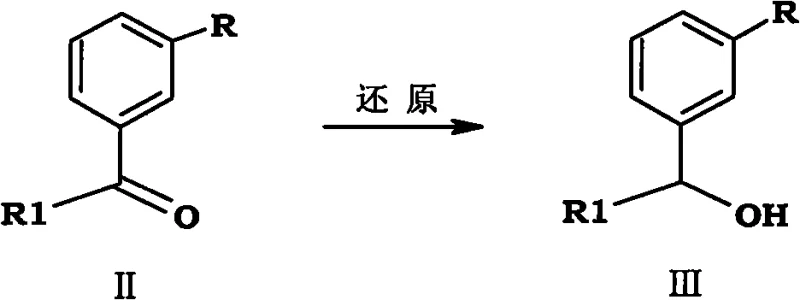
The core chemical innovation of this synthesis lies in the construction of the carbon skeleton via the alkylation of diethyl malonate with a halogenated intermediate derived from the initial ketone reduction. This nucleophilic substitution allows for the precise introduction of the ethyl and methyl side chains necessary for the Tapentadol structure without the stereochemical ambiguity often associated with Grignard additions to ketones. The subsequent hydrolysis and decarboxylation steps cleanly remove the ester protecting groups, yielding the desired carboxylic acid intermediate with high fidelity and minimal impurity carryover. Following acylation and amination, the final reduction step converts the amide to the amine using standard reducing agents like lithium aluminum hydride under controlled conditions, ensuring complete conversion without over-reduction of the aromatic ring. This stepwise assembly provides chemists with multiple opportunities for in-process quality control, allowing for the removal of impurities before they propagate through the synthesis, which is critical for meeting stringent regulatory specifications. The robustness of this mechanism ensures that the process remains stable even when scaled from laboratory benchtop to multi-ton reactor vessels.
Impurity control is meticulously managed through the selection of chiral resolving agents that selectively crystallize the desired (1R, 2R) enantiomer from the racemic mixture. The use of L-(-)-dibenzoyl tartaric acid creates a diastereomeric salt that exhibits distinct solubility properties, enabling efficient separation through simple filtration and recrystallization techniques rather than expensive preparative HPLC. This resolution strategy is complemented by the high purity of the precursors generated in the earlier malonate alkylation steps, which limits the formation of structurally related impurities that could co-crystallize with the product. Rigorous monitoring of reaction parameters such as pH during the hydrolysis and acidification stages further prevents the formation of polymeric byproducts or hydrolysis side-products that could compromise the final drug substance quality. By embedding these purification logic points directly into the synthetic design, the process inherently drives the impurity profile down to levels well below ICH Q3 guidelines. This level of control is paramount for R&D Directors who must validate the safety and efficacy of the drug substance before clinical or commercial release.
How to Synthesize Tapentadol Hydrochloride Efficiently
Executing this synthesis requires a disciplined approach to reaction monitoring and parameter control, beginning with the reduction of the starting ketone using sodium borohydride in a methanol or ethanol solvent system at controlled low temperatures. The subsequent halogenation step must be carefully managed to prevent over-halogenation or elimination side reactions, typically utilizing phosphorus tribromide or thionyl chloride under inert atmosphere conditions to ensure high conversion to the alkyl halide. Detailed standardized synthetic steps see the guide below, which outlines the precise stoichiometry and workup procedures required to maintain consistency across batches. Operators must adhere strictly to the specified temperature ranges during the malonate alkylation and decarboxylation phases to maximize yield and minimize the generation of thermal impurities. The final resolution and salt formation steps demand precise pH control and solvent selection to optimize the crystal habit and purity of the final hydrochloride salt. Adherence to these protocols ensures that the theoretical advantages of the patent are fully realized in practical production environments.
- Reduce 1-(3-substituted-phenyl)-1-ketone to the corresponding alcohol using sodium borohydride under mild alkaline conditions.
- Convert the alcohol to a halide using phosphorus tribromide or thionyl chloride, followed by nucleophilic substitution with diethyl malonate.
- Perform hydrolysis, decarboxylation, acylation, amination, and reduction, concluding with chiral resolution using L-(-)-dibenzoyl tartaric acid.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthetic route offers profound advantages that directly address the pain points of procurement managers and supply chain heads responsible for securing active pharmaceutical ingredients. The elimination of high-pressure hydrogenation and moisture-sensitive Grignard reagents drastically simplifies the equipment requirements, allowing production to occur in standard glass-lined or stainless steel reactors without the need for specialized high-pressure autoclaves. This reduction in capital intensity translates to lower depreciation costs per kilogram of product, enabling more competitive pricing structures for long-term supply agreements without compromising margin integrity. Furthermore, the use of common, non-proprietary starting materials mitigates the risk of supply disruptions caused by single-source vendor dependencies, enhancing the overall resilience of the procurement portfolio. The simplified purification workflow also reduces solvent consumption and waste disposal volumes, contributing to substantial cost savings in environmental compliance and waste management operations. These factors collectively create a more agile and cost-efficient supply chain capable of responding rapidly to fluctuations in market demand for analgesic medications.
- Cost Reduction in Manufacturing: The avoidance of expensive noble metal catalysts and complex chromatographic separation media significantly lowers the direct material costs associated with each production batch. By utilizing commodity chemicals like diethyl malonate and sodium borohydride, the process leverages economies of scale that are not available to routes relying on bespoke or hazardous reagents. The simplified workup procedures reduce labor hours and utility consumption, further driving down the conversion costs embedded in the final product price. Additionally, the higher overall yield achieved through minimized side reactions means less raw material is wasted, optimizing the atom economy of the entire manufacturing process. These cumulative efficiencies allow for a more aggressive pricing strategy while maintaining healthy profit margins for the manufacturer.
- Enhanced Supply Chain Reliability: The reliance on widely available raw materials ensures that production schedules are not held hostage by the lead times of niche chemical suppliers, providing a stable foundation for long-term planning. The robustness of the reaction conditions means that batch-to-batch variability is minimized, reducing the frequency of out-of-specification results that can delay shipments to customers. This consistency builds trust with downstream partners who rely on just-in-time delivery models to manage their own inventory levels effectively. Moreover, the safety improvements inherent in the process reduce the likelihood of unplanned shutdowns due to safety incidents, ensuring continuous operation and steady output. Such reliability is a critical differentiator in a market where supply continuity is often valued as highly as price.
- Scalability and Environmental Compliance: The process is designed with scale-up in mind, utilizing unit operations that are easily transferred from pilot plants to full-scale commercial facilities without significant re-engineering. The adherence to ICH residual solvent guidelines simplifies the regulatory filing process, accelerating the time to market for generic or new drug applications utilizing this intermediate. Reduced waste generation and the absence of heavy metal catalysts lower the environmental footprint of the manufacturing site, aligning with corporate sustainability goals and regulatory expectations. This environmental stewardship not only mitigates regulatory risk but also enhances the brand reputation of the supplier among environmentally conscious pharmaceutical clients. Ultimately, the process offers a future-proof solution that can adapt to tightening environmental regulations without requiring costly retrofits.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this novel synthetic pathway for Tapentadol Hydrochloride. These insights are derived directly from the experimental data and comparative analysis presented in the patent literature, providing a factual basis for decision-making. Understanding these details helps stakeholders evaluate the feasibility of adopting this technology for their specific supply chain requirements. The answers reflect a balance between chemical precision and commercial practicality, ensuring relevance for both technical and business audiences. We encourage further discussion with our technical team to explore how these advantages can be tailored to your specific project needs.
Q: What are the primary advantages of this synthesis method over traditional Grignard routes?
A: This method eliminates the need for hazardous Grignard reagents and high-pressure hydrogenation, significantly improving operational safety and reducing equipment costs for industrial scale-up.
Q: How is chirality controlled in this novel synthetic pathway?
A: Chirality is established through a resolution step using L-(-)-dibenzoyl tartaric acid, ensuring high enantiomeric excess suitable for pharmaceutical grade requirements without complex chromatographic separation.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the reaction conditions are mild, utilizing common solvents and reagents that meet ICH residual solvent guidelines, making it highly adaptable for multi-ton manufacturing environments.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tapentadol Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of deploying advanced synthetic technologies to meet the evolving demands of the global pharmaceutical market. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of patents like CN102557851B are fully actualized in our manufacturing facilities. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Tapentadol Hydrochloride meets the highest international standards for safety and efficacy. Our commitment to technical excellence allows us to navigate the complexities of chiral resolution and impurity control with precision, delivering a product that supports the therapeutic goals of our partners. By choosing us, you gain access to a supply chain that is both robust and responsive, capable of adapting to your volume requirements without compromising on quality.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis can drive value for your organization. Request a Customized Cost-Saving Analysis to quantify the potential economic benefits of switching to this superior manufacturing route. Our experts are ready to provide specific COA data and route feasibility assessments to support your internal validation processes. Let us collaborate to engineer a supply solution that enhances your competitive position in the analgesic sector while ensuring uninterrupted access to high-quality intermediates. Contact us today to initiate a conversation about your next project requirements.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →