Advanced Manufacturing of Chiral Bicyclic Amino Acid Esters for Antiviral APIs
The pharmaceutical industry's relentless pursuit of effective antiviral therapies has placed significant demand on the supply chain for high-quality chiral intermediates, particularly those serving as key structural fragments for next-generation influenza treatments. Patent CN111454166B discloses a groundbreaking methodology for the preparation of (2S,3S)-3-amino-bicyclo[2.2.2]octane-2-carboxylate, a critical building block for the synthesis of Pimodivir, a potent influenza virus RNA polymerase inhibitor currently in advanced clinical development. This technical disclosure represents a paradigm shift from hazardous, low-yielding legacy processes to a streamlined, safety-oriented synthetic strategy that leverages chiral reductive amination and stereoselective inversion. For global procurement leaders and R&D directors, understanding the nuances of this patented route is essential, as it offers a viable pathway to secure reliable supplies of this complex bicyclic amino acid derivative while mitigating the substantial risks associated with traditional azide chemistry and cryogenic reactions.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of this rigid bicyclic scaffold has been plagued by severe operational hazards and economic inefficiencies that hinder commercial viability. As illustrated in prior art documentation, early routes often relied on the Diels-Alder reaction followed by a Curtius rearrangement to install the amine functionality. This approach necessitates the generation and handling of acyl azides, which are notoriously unstable and explosive, posing unacceptable safety risks for multi-ton manufacturing environments. Furthermore, alternative pathways utilizing Michael additions with chiral amino anions require stringent cryogenic conditions and expensive metal catalysts, driving up utility costs and limiting reactor throughput. Other reported methods involving nitro-alkene intermediates introduce toxic nitromethane and unstable nitro groups into the process stream, creating significant waste disposal challenges and potential regulatory hurdles for environmental compliance.
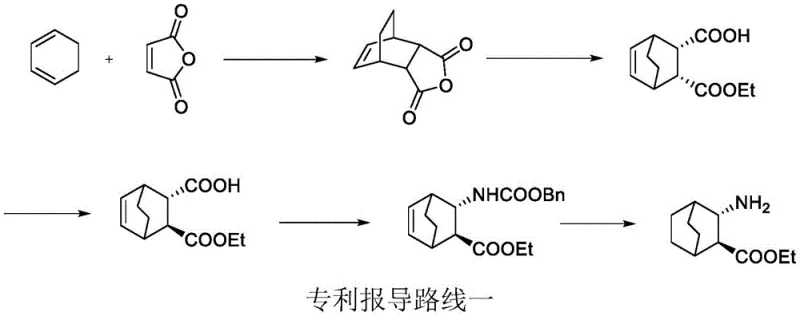
The Novel Approach
In stark contrast to these perilous legacy methods, the innovative process detailed in the patent utilizes a remarkably elegant sequence starting from 3-carbonyl-bicyclo[2.2.2]octane-2-carboxylate. By employing a chiral reductive amination strategy, the synthesis bypasses the need for dangerous azide intermediates entirely. The route capitalizes on the steric differentiation provided by chiral amines, such as S-1-phenylethylamine or S-1-naphthylethylamine, to direct the initial stereochemical outcome. This is followed by a controlled base-mediated epimerization that inverts the ester configuration to the desired thermodynamic state. The elimination of explosive reagents and the avoidance of ultra-low temperature operations fundamentally transform the risk profile of the manufacturing process, offering a robust platform for the cost reduction in pharmaceutical intermediate manufacturing that supply chain managers desperately seek.
Mechanistic Insights into Chiral Reductive Amination and Epimerization
The core of this technological breakthrough lies in the precise control of stereochemistry through a tandem reductive amination and inversion sequence. The process initiates with the condensation of the ketone starting material with a chiral primary amine under acidic catalysis, forming an enamine intermediate that locks the nitrogen functionality onto the bicyclic framework. Subsequent reduction, preferably using sodium triacetoxyborohydride or catalytic hydrogenation over platinum or palladium, occurs with high facial selectivity dictated by the chiral auxiliary. This step establishes the initial (2R,3S) configuration with excellent diastereomeric excess. The true ingenuity of the method is revealed in the subsequent treatment with a strong non-nucleophilic base, such as sodium tert-butoxide. This reagent facilitates the deprotonation of the alpha-proton adjacent to the ester, generating an enolate that allows for thermodynamic equilibration. Due to the steric constraints of the bicyclic system and the influence of the adjacent amino group, the equilibrium favors the formation of the (2S,3S) diastereomer, effectively inverting the stereocenter to match the natural configuration required for biological activity.
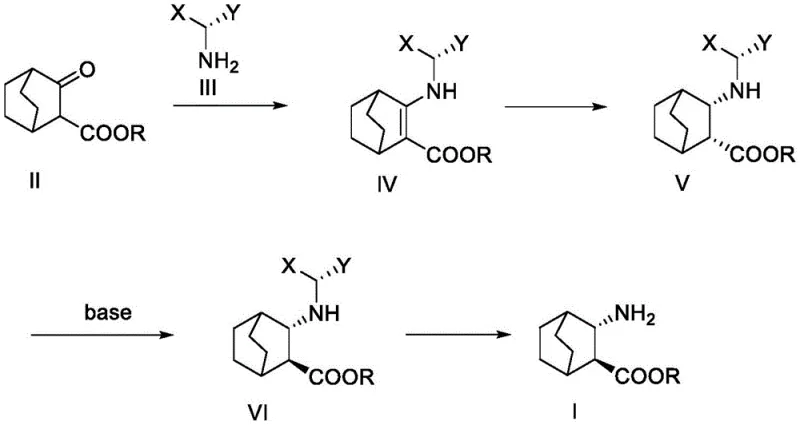
From an impurity control perspective, this mechanism offers distinct advantages over random racemic syntheses followed by resolution. Because the chirality is induced early via the chiral amine auxiliary and then locked in through thermodynamic control, the formation of unwanted diastereomers is minimized throughout the sequence. The patent data highlights that the intermediate compounds, specifically the protected amino esters designated as Formula V and Formula VI, can be isolated with high purity. This inherent selectivity reduces the burden on downstream purification units, such as chromatography columns or repeated recrystallizations, which are often the bottleneck in fine chemical production. The ability to achieve a final chiral purity exceeding 99.5% after simple workup demonstrates the robustness of this mechanistic design, ensuring that the final API meets the rigorous specifications demanded by global regulatory bodies for antiviral medications.
How to Synthesize (2S,3S)-3-amino-bicyclo[2.2.2]octane-2-carboxylate Efficiently
The practical execution of this synthesis involves a logical progression of unit operations that are well-suited for standard chemical processing equipment. The protocol begins with the formation of the enamine in a refluxing solvent like toluene, followed by an in-situ or isolated reduction step. The critical inversion step requires careful temperature control during the addition of the strong base to prevent side reactions, typically conducted in tetrahydrofuran or tert-butanol mixtures. Finally, the removal of the chiral auxiliary is achieved through standard catalytic hydrogenolysis, a ubiquitous operation in pharmaceutical plants.
- Condense 3-carbonyl-bicyclo[2.2.2]octane-2-carboxylate with a chiral amine (e.g., S-1-phenylethylamine) under acidic conditions to form an enamine intermediate.
- Perform stereoselective reduction of the enamine double bond using a reducing agent like sodium triacetoxyborohydride or catalytic hydrogenation to establish the (2R,3S) configuration.
- Treat the reduced intermediate with a strong base such as sodium tert-butoxide to induce epimerization at the alpha-position, inverting the ester configuration to (2S,3S).
- Execute catalytic hydrogenation using palladium on carbon to remove the chiral auxiliary protecting group, yielding the final target amino ester.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this novel synthetic route translates directly into enhanced supply security and optimized cost structures. The elimination of hazardous reagents like diphenylphosphoryl azide removes the need for specialized blast-proof facilities and expensive safety protocols, which inherently lowers the overhead cost of production. Furthermore, the reliance on commodity chemicals such as ethyl 3-carbonyl-bicyclo[2.2.2]octane-2-carboxylate and common chiral amines ensures a stable raw material supply chain that is less susceptible to the volatility seen with exotic catalysts or unstable precursors.
- Cost Reduction in Manufacturing: The process achieves a total yield of more than 65%, which is a substantial improvement over the less than 20% yields reported for older routes. This dramatic increase in efficiency means that less raw material is wasted per kilogram of finished product, directly lowering the cost of goods sold. Additionally, the avoidance of cryogenic conditions reduces energy consumption significantly, as the reactions proceed under mild thermal conditions ranging from room temperature to moderate reflux, eliminating the need for expensive cooling infrastructure.
- Enhanced Supply Chain Reliability: By removing the dependency on explosive azides and unstable nitro compounds, the manufacturing timeline becomes more predictable and less prone to safety-related shutdowns. The robustness of the reductive amination and hydrogenation steps allows for continuous processing or large batch campaigns without the frequent interruptions associated with handling high-risk intermediates. This reliability is crucial for maintaining the continuity of supply for critical antiviral drugs, especially during pandemic scenarios where demand spikes unexpectedly.
- Scalability and Environmental Compliance: The synthetic route is designed with green chemistry principles in mind, avoiding the generation of heavy metal waste or toxic nitrogenous byproducts typical of Curtius rearrangements. The use of standard solvents like ethanol, ethyl acetate, and toluene simplifies solvent recovery and recycling processes. This environmental compatibility not only reduces waste disposal costs but also aligns with the increasingly stringent sustainability mandates imposed by global health authorities and corporate ESG goals, facilitating smoother regulatory approvals for commercial scale-up.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent literature, offering a transparent view of the process capabilities.
Q: What are the safety advantages of this new synthesis route compared to traditional methods?
A: Unlike conventional routes that rely on hazardous Curtius rearrangements involving explosive azides or unstable nitro compounds, this novel process utilizes mild reductive amination and standard hydrogenation, significantly eliminating high-energy intermediates and enhancing operational safety for large-scale production.
Q: What is the expected chiral purity of the final product using this method?
A: The process is designed to achieve high stereocontrol through the use of chiral amines and base-catalyzed inversion. The patent data indicates that the target product can reach a chiral purity of greater than 99.5% following simple crystallization and purification steps.
Q: Is this process suitable for industrial scale-up?
A: Yes, the route avoids extreme conditions such as ultra-low temperatures required in previous methods. It employs readily available raw materials and robust reaction conditions, making it highly adaptable for commercial scale-up from kilogram to multi-ton quantities without specialized equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (2S,3S)-3-amino-bicyclo[2.2.2]octane-2-carboxylate Supplier
As the global demand for next-generation influenza therapeutics continues to rise, securing a dependable source of high-purity chiral intermediates is paramount for pharmaceutical developers. NINGBO INNO PHARMCHEM stands at the forefront of this sector, leveraging deep expertise in asymmetric synthesis to deliver complex molecules with unparalleled consistency. Our facility is equipped with extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements whether you are in the pre-clinical phase or preparing for market launch. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of (2S,3S)-3-amino-bicyclo[2.2.2]octane-2-carboxylate meets the exacting standards required for API synthesis.
We invite you to collaborate with our technical team to explore how this advanced manufacturing route can optimize your supply chain. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific project needs. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our innovative processes can drive value and efficiency in your antiviral drug development programs.