Optimizing PARP Inhibitor Production: Advanced Rucaparib Intermediate Synthesis and Commercial Scale-Up
The pharmaceutical landscape for oncology treatments continues to evolve rapidly, with Poly (ADP-ribose) polymerase (PARP) inhibitors standing out as a critical class of chemotherapeutic agents for treating advanced ovarian cancer and BRCA-mutated malignancies. Within this high-stakes domain, the efficiency and safety of the supply chain for key intermediates like Rucaparib (also known as Lucapanib) are paramount for global drug manufacturers. A recent technological breakthrough, documented in patent CN111217818A, introduces a transformative synthesis method that addresses long-standing bottlenecks in the production of these tricyclic compounds. This innovation not only streamlines the chemical pathway but also fundamentally alters the safety profile and economic feasibility of large-scale manufacturing. By shifting away from hazardous reducing agents and complex purification sequences, this new methodology offers a robust framework for producing high-purity pharmaceutical intermediates. For procurement leaders and technical directors, understanding the nuances of this patent is essential for securing a competitive edge in the API market. The following analysis dissects the technical merits and commercial implications of this advanced synthesis route, providing a clear roadmap for integrating these improvements into your supply chain strategy.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Rucaparib and its related tricyclic structures has been plagued by inefficient multi-step processes that rely on hazardous reagents and extensive purification protocols. The conventional route, often referenced in earlier intellectual property such as U.S. Patent No. 6,977,298, typically involves the reduction of imine intermediates using sodium cyanoborohydride (NaBH3CN). This specific reagent poses significant safety risks due to the potential generation of hydrogen cyanide (HCN) gas, a highly toxic byproduct that necessitates stringent containment measures and specialized waste treatment facilities. Furthermore, the traditional pathway requires the isolation and purification of multiple intermediate salts, such as the hydrochloride form, which introduces substantial material loss at each stage. These cumulative losses result in an overall yield that often hovers around a mere 31%, making the process economically burdensome and environmentally taxing. The need for neutralization steps using potassium hydroxide and methanol further complicates the workflow, increasing solvent consumption and processing time. For supply chain managers, these inefficiencies translate into higher costs, longer lead times, and increased regulatory scrutiny regarding hazardous waste disposal.
The Novel Approach
In stark contrast to the legacy methods, the novel approach disclosed in CN111217818A leverages a streamlined catalytic hydrogenation strategy that bypasses the need for toxic cyanoborohydrides entirely. This innovative route converts the aldehyde precursor directly into an imine intermediate using methylamine, which is then immediately subjected to hydrogenation in the presence of a palladium on carbon (Pd/C) catalyst. This telescoping of steps eliminates the isolation of the imine intermediate, thereby reducing handling losses and simplifying the reactor workflow. The use of catalytic hydrogenation is not only safer but also significantly more atom-economical, utilizing hydrogen gas as the reducing agent and producing no toxic gaseous byproducts. Experimental embodiments within the patent demonstrate that this method can achieve yields ranging from 70% to as high as 84%, representing a dramatic improvement over the conventional 31% benchmark. By removing the need for salt formation and subsequent neutralization, the process reduces solvent usage and waste generation, aligning perfectly with modern green chemistry principles. This shift represents a paradigm change in how complex tricyclic PARP inhibitors are manufactured, offering a scalable and sustainable alternative for industrial production.
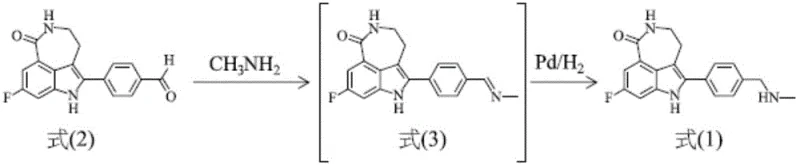
Mechanistic Insights into Pd/C-Catalyzed Hydrogenation
The core of this technological advancement lies in the precise manipulation of the reduction mechanism, specifically the transition from an imine intermediate to the final amine product. In the novel pathway, the aldehyde compound of Formula (II) reacts with methylamine to form the imine compound of Formula (III). Unlike traditional methods that would isolate this imine or reduce it with stoichiometric hydride sources, this process keeps the imine in solution for immediate catalytic hydrogenation. The palladium catalyst facilitates the addition of hydrogen across the carbon-nitrogen double bond of the imine, effectively saturating the molecule to form the desired tricyclic amine of Formula (I). This catalytic cycle is highly efficient and can be tuned by adjusting parameters such as catalyst loading (e.g., 5% to 10% Pd/C) and hydrogen pressure. The ability to perform this reaction without isolating the imine is crucial, as imines can sometimes be unstable or prone to hydrolysis. By maintaining a continuous flow from imine formation to reduction, the process minimizes exposure to potentially degrading conditions and maximizes the throughput of the reactor. This mechanistic elegance ensures that the stereochemistry and structural integrity of the complex tricyclic core are preserved throughout the transformation.
Beyond the primary reduction step, the control of impurities is a critical aspect of this mechanism that directly impacts the quality of the final API. The use of catalytic hydrogenation inherently reduces the formation of side products associated with hydride reductions, such as over-reduced species or boron-containing impurities that are difficult to remove. The patent data indicates that the crude product obtained from this hydrogenation step is of sufficient purity to proceed to subsequent salt formation without extensive chromatographic purification. This is a significant advantage for R&D directors focused on impurity profiles, as it simplifies the downstream processing and reduces the risk of genotoxic impurities carrying over into the final drug substance. Furthermore, the specific crystallization of the (S)-camphorsulfonate salt provides an additional purification checkpoint, leveraging the distinct solubility properties of the chiral salt to exclude remaining trace impurities. The combination of a clean catalytic reaction and a robust crystallization process ensures that the final material meets the stringent purity specifications required for oncology therapeutics.
How to Synthesize Rucaparib Efficiently
The implementation of this synthesis route requires a disciplined approach to reaction conditions and reagent quality to fully realize the yield and safety benefits described in the patent. The process begins with the preparation of the aldehyde precursor via Suzuki coupling, followed by the critical imine formation and hydrogenation sequence. Operators must ensure that the methylamine addition is controlled to prevent exotherms and that the hydrogenation reactor is properly purged and pressurized to maintain catalyst activity. The elimination of isolation steps means that in-process controls (IPC) become even more vital to monitor the conversion of the aldehyde to the imine and subsequently to the amine. Detailed standard operating procedures (SOPs) should reflect the continuous nature of this process, emphasizing the transfer of reaction mixtures between stages without exposure to atmospheric moisture which could hydrolyze the imine. The following guide outlines the standardized synthesis steps derived from the patent embodiments to ensure reproducibility and safety in a commercial setting.
- Perform Suzuki coupling between halogenated tricyclic precursors and formylphenylboronic acid to construct the core biaryl structure.
- Convert the resulting aldehyde intermediate into an imine derivative using methylamine under controlled temperature conditions.
- Execute catalytic hydrogenation using Pd/C to reduce the imine directly to the final amine without isolating toxic intermediates.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthesis route offers tangible benefits that extend far beyond simple chemical efficiency. The primary advantage lies in the drastic simplification of the manufacturing workflow, which directly correlates to reduced operational expenditures and enhanced supply reliability. By eliminating the use of sodium cyanoborohydride, facilities can avoid the high costs associated with handling hazardous materials, including specialized storage, personal protective equipment, and toxic waste disposal fees. This removal of a significant safety hazard also reduces the regulatory burden and insurance premiums associated with the production site. Furthermore, the increase in overall yield from approximately 31% to over 70% means that less raw material is required to produce the same amount of final API, leading to substantial cost savings on starting materials. The reduction in processing steps also shortens the manufacturing cycle time, allowing for faster turnaround on orders and improved responsiveness to market demand fluctuations. These factors combine to create a more resilient and cost-effective supply chain for PARP inhibitor intermediates.
- Cost Reduction in Manufacturing: The economic impact of switching to this catalytic hydrogenation route is profound, driven primarily by the elimination of expensive and hazardous reagents. Traditional methods rely on stoichiometric amounts of reducing agents that generate significant waste, whereas the new method uses catalytic amounts of palladium and hydrogen gas, which are far more cost-effective on a per-kilogram basis. Additionally, the removal of intermediate isolation and purification steps reduces the consumption of solvents and energy, further driving down the variable cost of production. The higher yield ensures that the fixed costs of the facility are amortized over a larger output of saleable product, improving the overall margin structure. These savings can be passed down the supply chain, offering competitive pricing for the final pharmaceutical intermediate without compromising on quality or compliance standards.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by the complexity of synthesis routes that depend on hard-to-source reagents or fragile intermediates. This new method utilizes widely available starting materials and robust catalysts that are standard in the fine chemical industry, reducing the risk of supply disruptions. The telescoping of steps minimizes the number of hand-offs between different processing units or external vendors, thereby reducing the logistical friction and potential for delays. The stability of the process also means that batch-to-batch variability is minimized, ensuring consistent quality and delivery schedules for downstream API manufacturers. For supply chain heads, this reliability translates into lower safety stock requirements and a more predictable inventory management strategy, ultimately strengthening the partnership between the intermediate supplier and the drug developer.
- Scalability and Environmental Compliance: Scaling chemical processes from the lab to commercial production often reveals hidden bottlenecks, particularly regarding heat transfer and waste management. The catalytic hydrogenation described here is inherently scalable, as hydrogenation reactors are standard equipment in most GMP facilities and the reaction exotherm is manageable. The absence of toxic HCN gas generation simplifies the environmental compliance landscape, making it easier to obtain permits for expansion or operation in regions with strict environmental regulations. The reduced solvent load and waste generation align with sustainability goals, which are increasingly becoming a criterion for supplier selection by major pharmaceutical companies. This environmental stewardship not only mitigates regulatory risk but also enhances the brand reputation of the manufacturing partner as a responsible and forward-thinking entity in the global healthcare ecosystem.
Frequently Asked Questions (FAQ)
The transition to a new synthesis platform often raises technical and commercial questions among stakeholders who are accustomed to legacy methods. The following answers are derived directly from the technical specifications and experimental data provided in the patent documentation to address common concerns regarding safety, yield, and quality. Understanding these details is crucial for making informed decisions about supplier qualification and process validation. The data reflects a rigorous testing regime that confirms the robustness of the new method under various conditions, providing confidence in its applicability for commercial scale-up. These insights are intended to clarify the operational realities of implementing this technology within an existing manufacturing framework.
Q: How does the new catalytic hydrogenation route improve safety compared to conventional methods?
A: The novel process eliminates the use of sodium cyanoborohydride (NaBH3CN), thereby preventing the generation of toxic hydrogen cyanide (HCN) gas during the reduction step, significantly enhancing operational safety for mass production.
Q: What yield improvements can be expected with this patented synthesis route?
A: Experimental data indicates that the new route achieves yields exceeding 70%, with optimized conditions reaching up to 84%, compared to approximately 31% yield in conventional multi-step purification routes.
Q: Is the crystalline form of the camphorsulfonate salt stable for long-term storage?
A: Yes, the specific crystalline form of Rucaparib (S)-camphorsulfonate described exhibits a high melting point of approximately 304°C and distinct XRD characteristics, ensuring excellent thermal stability and shelf-life for pharmaceutical applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Rucaparib Supplier
The technical potential of this patented synthesis route is immense, but realizing its value requires a manufacturing partner with the expertise to execute complex chemistry at scale. NINGBO INNO PHARMCHEM stands as a premier CDMO expert, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facilities are equipped with state-of-the-art hydrogenation reactors and stringent purity specifications are maintained through our rigorous QC labs, ensuring that every batch of Rucaparib intermediate meets the highest global standards. We understand the critical nature of oncology supply chains and are committed to delivering consistency, quality, and reliability. Our team is well-versed in the nuances of PARP inhibitor synthesis and can seamlessly integrate this novel catalytic hydrogenation technology into our production lines to serve your specific volume requirements.
We invite you to engage with us to explore how this advanced manufacturing route can optimize your procurement strategy and reduce your overall cost of goods. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your project's specific needs, demonstrating the tangible economic benefits of switching to this safer and more efficient process. We encourage you to contact us to request specific COA data and route feasibility assessments that will validate the compatibility of this intermediate with your downstream API synthesis. By partnering with us, you gain access to a supply chain that is not only cost-effective but also resilient and compliant with the evolving regulatory landscape of the pharmaceutical industry.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →