Advanced Synthesis of Relugolix Intermediate Impurity VI for Pharmaceutical Quality Control
The pharmaceutical industry faces increasing regulatory scrutiny regarding the purity profiles of active pharmaceutical ingredients (APIs), particularly for potent molecules like Relugolix, a gonadotropin-releasing hormone (GnRH) antagonist used in treating uterine fibroids and prostate cancer. Patent CN114380810A addresses a critical gap in the quality control infrastructure for this drug by disclosing a novel method for the preparation of a specific Relugolix intermediate impurity, designated as Formula VI. Historically, the lack of authenticated reference standards for such impurities has hindered accurate quantitative analysis, posing risks to batch release and patient safety. This patent introduces a robust, five-step synthetic pathway that transforms a precursor compound (Compound I) into the target impurity through a sequence of hydrolysis, condensation, reduction, cyclization, and ring-opening reactions. By enabling the production of this impurity with purity levels exceeding 98% and yields suitable for gram-scale production, the technology provides QC laboratories with the essential tools needed to validate analytical methods and ensure the highest standards of drug safety.
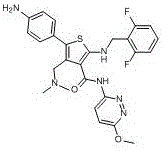
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the identification and quantification of process-related impurities in complex small molecule drugs like Relugolix have relied heavily on isolating these byproducts directly from crude reaction mixtures or final API batches. This approach is fraught with significant technical and logistical challenges, primarily because impurities are often generated in trace quantities, making their isolation labor-intensive and economically unviable. Furthermore, impurities isolated from process streams may not be chemically stable or may co-elute with other degradation products, leading to ambiguous structural characterization. For procurement and supply chain managers, relying on isolation means that reference standards are unavailable until large-scale API production has already commenced, creating a bottleneck in method validation and regulatory filing. The inability to procure authentic samples of specific structural variants, such as the ring-opened or cyclized byproducts described in this patent, leaves manufacturers vulnerable to unexpected regulatory queries regarding impurity thresholds and toxicological assessments.
The Novel Approach
The methodology disclosed in patent CN114380810A represents a paradigm shift from passive isolation to active, deliberate synthesis of the target impurity. Instead of scavenging for trace byproducts, this approach utilizes a defined starting material, Compound I, and subjects it to a controlled series of chemical transformations designed to mimic the specific structural deviations found in the final drug substance. This proactive strategy allows for the generation of the impurity (Formula VI) in substantial quantities, independent of the main API production schedule. The process is optimized to deliver high purity, with reported HPLC purity values reaching up to 99.78% in specific examples, which is critical for its use as a primary reference standard. For R&D directors, this means having immediate access to well-characterized materials that can be used to spike samples, determine detection limits, and validate cleaning procedures, thereby de-risking the entire development timeline and ensuring a smoother path to commercial approval.
Mechanistic Insights into the Multi-Step Synthetic Route
The synthesis of the Relugolix intermediate impurity involves a sophisticated orchestration of organic transformations, beginning with the hydrolysis of an ester or similar functionality in Compound I to generate the carboxylic acid intermediate, Compound II. This initial step typically employs mild alkaline conditions, such as sodium hydroxide in ethanol or methanol at temperatures ranging from 45°C to 55°C, ensuring selective cleavage without damaging sensitive functional groups elsewhere in the molecule. Following isolation, Compound II undergoes a condensation reaction with 3-amino-6-methoxypyridazine, a key building block that introduces the nitrogenous heterocycle essential for the biological activity of the parent drug. This coupling is facilitated by modern peptide coupling agents like 1-propylphosphonic anhydride (T3P) or carbodiimides in the presence of an acid scavenger like N,N-diisopropylethylamine, proceeding efficiently at moderate temperatures (35-45°C) to yield the amide-linked Compound III with high fidelity.
The subsequent transformation involves the reduction of a nitro group present on the aromatic ring of Compound III to an aniline derivative (Compound IV), a critical step that enables the downstream cyclization. This reduction is achieved using catalytic hydrogenation with palladium on carbon under controlled hydrogen pressure (1.5-2.0 MPa), a method chosen for its cleanliness and scalability compared to stoichiometric metal reductions. Once the amine is generated, an intramolecular cyclization is triggered under basic conditions using potassium carbonate and a phase transfer catalyst like tetrabutylammonium bromide in tetrahydrofuran, forming the fused ring system of Compound V. The final step involves a ring-opening reaction using sodium methoxide in methanol at elevated temperatures (55-70°C), which rearranges the molecular skeleton to match the specific topology of the target impurity Formula VI. This comprehensive pathway demonstrates a deep understanding of reactivity patterns, allowing for the precise construction of complex impurity structures.
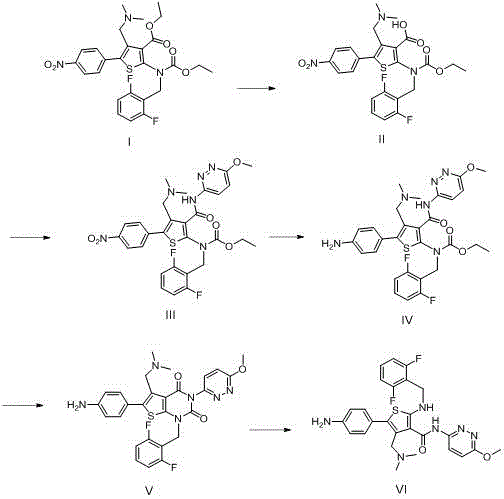
How to Synthesize Relugolix Intermediate Impurity VI Efficiently
The successful execution of this synthesis requires strict adherence to the reaction parameters outlined in the patent to ensure the formation of the correct regioisomer and to minimize the generation of secondary byproducts. The process is designed to be operationally simple, utilizing common organic solvents and reagents that are readily available in standard pharmaceutical manufacturing facilities. Detailed below is the strategic overview of the standardized synthesis protocol, which balances reaction kinetics with product stability to maximize overall yield. For laboratory technicians and process chemists looking to implement this route, attention to the specific workup procedures, such as pH adjustments and extraction protocols, is vital to achieving the reported purity levels of >98%.
- Hydrolyze Compound I using alkali in alcohol solvent at 45-55°C to obtain Compound II.
- Condense Compound II with 3-amino-6-methoxypyridazine using a coupling agent like T3P to form Compound III.
- Reduce the nitro group of Compound III using Pd/C catalyst under hydrogen pressure (1.5-2.0 MPa) to yield Compound IV.
- Perform intramolecular cyclization of Compound IV using potassium carbonate and a phase transfer catalyst to generate Compound V.
- Execute ring-opening of Compound V using sodium methoxide in methanol to finalize the Relugolix intermediate impurity (Formula VI).
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the ability to synthesize specific impurities like Formula VI offers profound advantages for supply chain resilience and cost management in pharmaceutical manufacturing. By decoupling the production of reference standards from the main API campaign, companies can avoid the costly delays associated with waiting for pilot plant runs to generate sufficient material for QC testing. This independence allows for parallel processing, where analytical method validation can proceed concurrently with process development, significantly compressing the overall time-to-market for the generic or branded product. Furthermore, having a reliable internal or contracted source for these critical materials mitigates the risk of supply disruptions that could otherwise halt batch release or regulatory submissions.
- Cost Reduction in Manufacturing: The implementation of this synthetic route eliminates the need for expensive and inefficient isolation processes that typically yield negligible amounts of impurity from tons of crude product. By using a directed synthesis with high-yielding steps (reported yields of 90-95% in key stages), the cost per gram of the reference standard is drastically reduced compared to traditional isolation methods. Additionally, the use of commodity chemicals and standard catalysts like palladium on carbon ensures that raw material costs remain predictable and low, avoiding the premium pricing often associated with custom-synthesized specialty compounds.
- Enhanced Supply Chain Reliability: Establishing a defined synthetic pathway ensures a consistent and reproducible supply of the impurity standard, which is crucial for long-term stability studies and ongoing quality monitoring throughout the product lifecycle. Unlike isolation, which is dependent on the variability of the main reaction's impurity profile, this synthesis provides a steady stream of material regardless of fluctuations in the primary API manufacturing process. This reliability empowers procurement managers to secure long-term contracts for reference materials with confidence, knowing that lead times will be short and supply continuity is guaranteed by the robustness of the chemical route.
- Scalability and Environmental Compliance: The process utilizes solvents such as ethanol, ethyl acetate, and dichloromethane, which are well-understood in terms of waste management and recycling, facilitating easier environmental compliance during scale-up. The reaction conditions, including moderate temperatures and pressures, are compatible with standard glass-lined or stainless steel reactors, allowing for seamless translation from gram-scale laboratory synthesis to kilogram-scale production without the need for specialized equipment. This scalability ensures that as the demand for Relugolix grows, the capacity to produce the necessary quality control materials can expand in tandem without requiring significant capital investment in new infrastructure.
Frequently Asked Questions (FAQ)
The following questions address common technical and operational inquiries regarding the synthesis and application of Relugolix intermediate impurities. These insights are derived directly from the experimental data and technical specifications provided in the patent literature, offering clarity on the feasibility and benefits of this approach for pharmaceutical stakeholders.
Q: Why is synthesizing Relugolix impurities necessary for API quality control?
A: Natural impurities found in crude API batches are often present in trace amounts and are difficult to isolate in sufficient quantities for structural confirmation or method validation. Synthetic preparation ensures a stable, high-purity supply of reference standards needed for accurate HPLC quantification and regulatory compliance.
Q: What are the critical reaction conditions for the reduction step in this process?
A: The reduction of the nitro intermediate (Compound III) requires precise control of hydrogen pressure between 1.5-2.0 MPa using a palladium on carbon catalyst in dichloromethane. Maintaining these parameters ensures complete conversion to the amine (Compound IV) without over-reduction or side reactions.
Q: How does this synthetic route improve supply chain reliability for reference standards?
A: By establishing a robust multi-step synthesis starting from readily available precursors, manufacturers can produce gram-to-kilogram quantities of the impurity on demand. This eliminates the dependency on unpredictable isolation from process waste streams, ensuring consistent availability for QC laboratories.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Relugolix Impurity Supplier
At NINGBO INNO PHARMCHEM, we understand that the integrity of your pharmaceutical products relies heavily on the quality and availability of critical reference standards and intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that whether you need gram quantities for analytical validation or larger volumes for process optimization, we can deliver with precision. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify the identity and purity of every batch, guaranteeing that our materials meet the exacting demands of global regulatory agencies.
We invite you to collaborate with us to leverage this advanced synthetic technology for your Relugolix development programs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can support your supply chain goals and accelerate your project timelines.