Advanced Manufacturing of Fibrinogen Receptor Antagonist Intermediates via Optimized O-Alkylation
The pharmaceutical industry continuously seeks robust synthetic routes for complex bioactive molecules, particularly fibrinogen receptor antagonists which play a critical role in antithrombotic therapy. Patent CN1076441A discloses a groundbreaking high-efficiency synthesis method that significantly streamlines the production of these vital intermediates compared to prior art. Historically, the preparation of such compounds involved cumbersome eleven-step processes laden with hazardous reagents and low overall yields. This new methodology condenses the synthesis into a concise four-step sequence, leveraging innovative protection strategies and selective coupling reactions to achieve superior purity and scalability. By replacing dangerous sodium hydride protocols with safer aqueous base conditions and utilizing BSTFA for stereoselective protection, this process represents a paradigm shift in manufacturing feasibility. The following analysis details how this technology enables the production of high-purity pharmaceutical intermediates suitable for commercial scale-up.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic pathways for fibrinogen receptor antagonists have long been plagued by significant operational hazards and purification bottlenecks that hinder commercial viability. A primary drawback of conventional methods is the reliance on sodium hydride (NaH) in dimethylformamide (DMF) to induce ether formation, a combination known for its potential explosiveness and difficult handling characteristics on a large scale. Furthermore, these legacy processes often necessitate extensive chromatographic purification steps to isolate the desired product from complex reaction mixtures, which drastically increases production costs and solvent waste. The multi-step nature of previous approaches, sometimes exceeding eleven distinct operations, compounds these issues by accumulating yield losses at each stage and increasing the risk of stereochemical degradation. Such inefficiencies create substantial barriers for supply chain reliability, making it challenging to secure consistent volumes of high-quality active pharmaceutical ingredient (API) precursors for global markets.
The Novel Approach
The innovative route described in the patent overcomes these historical challenges through a cleverly designed sequence that prioritizes safety, selectivity, and ease of isolation. Central to this advancement is the use of bis(trimethylsilyl)trifluoroacetamide (BSTFA) to mediate the sulfonylation of tyrosine, which acts as a temporary protecting group to preserve chirality while enabling selective N-functionalization. Instead of hazardous metal hydrides, the ether linkage is formed using simple aqueous potassium hydroxide (3N KOH) in polar aprotic solvents like DMSO, conditions that are far more manageable in industrial reactors. Crucially, the process eliminates the need for column chromatography entirely by employing precise pH-controlled crystallization techniques to remove impurities. This shift from chromatographic purification to crystallization not only reduces solvent consumption but also facilitates the direct isolation of the product in high purity, demonstrating a clear path toward cost-effective manufacturing.
Mechanistic Insights into BSTFA-Mediated Sulfonylation and O-Alkylation
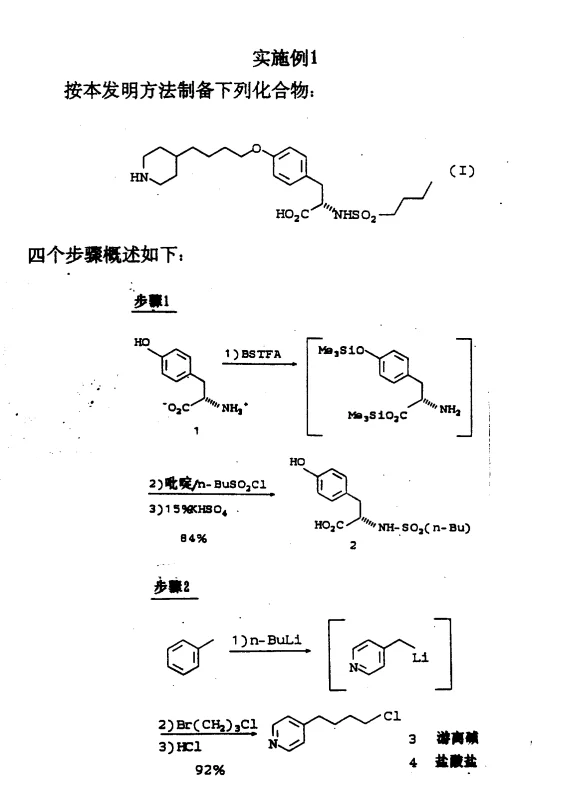
The chemical elegance of this synthesis lies in its ability to control reactivity and stereochemistry through specific reagent choices. In the initial sulfonylation step, BSTFA reacts with L-tyrosine to form a bis-O,O'-silylated intermediate, effectively masking the phenolic hydroxyl and carboxylic acid groups. This temporary protection is critical because it prevents unwanted side reactions and, more importantly, shields the alpha-carbon from racemization during the subsequent attack by the sulfonyl chloride. The reaction proceeds smoothly in acetonitrile with pyridine as a base, yielding the N-sulfonylated tyrosine derivative in excellent yields of approximately 84%. This mechanistic safeguard ensures that the final product retains the necessary L-configuration required for biological activity, a common failure point in less optimized synthetic routes.
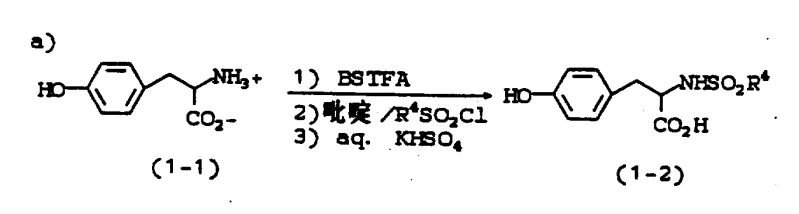
Following the preparation of the chloroalkyl pyridine fragment, the core carbon-oxygen bond is constructed via a nucleophilic substitution reaction. The phenolic oxygen of the protected tyrosine acts as the nucleophile, attacking the terminal chloride of the alkyl chain in the presence of 3N KOH. The choice of solvent plays a pivotal role here; while solvents like DMPU minimize dialkylation side products, DMSO is preferred for its balance of reactivity and cost. The reaction is conducted at elevated temperatures around 65°C to ensure complete conversion. Mechanistically, the strong base deprotonates the phenol to generate a phenoxide ion, which is highly reactive towards the primary alkyl halide. The process is designed to suppress the formation of dialkylated impurities, which are kept below 2% through careful control of stoichiometry and reaction time, ensuring a clean profile before the final hydrogenation step.
How to Synthesize Fibrinogen Receptor Antagonist Intermediate Efficiently
The synthesis of this high-value intermediate is achieved through a logical progression of four distinct chemical transformations that can be executed in standard stainless steel equipment. The process begins with the activation of L-tyrosine, followed by the independent preparation of the heterocyclic side chain, and culminates in their convergence and final reduction. Each step has been optimized to maximize yield and minimize impurity carryover, making it an ideal candidate for technology transfer. The detailed standardized synthesis steps are outlined below to guide process development teams in replicating these results.
- Perform BSTFA-mediated sulfonylation of L-tyrosine with n-BuSO2Cl to obtain N-sulfonylated tyrosine with high stereochemical integrity.
- Prepare 4-(4-pyridyl)butyl chloride via lithiation of 4-picoline followed by quenching with 3-bromo-1-chloropropane.
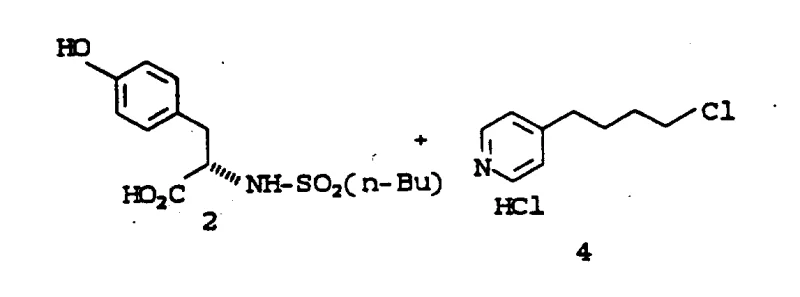
- Execute selective O-alkylation using 3N KOH in DMSO at 65°C to couple the tyrosine derivative with the chloroalkyl pyridine.
- Conduct selective hydrogenation of the pyridine ring using Pd/C in acetic acid to yield the final piperidine-containing antagonist.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible strategic benefits that extend beyond mere chemical yield. By fundamentally altering the reagent profile and purification strategy, the process addresses key pain points related to cost volatility, regulatory compliance, and production throughput. The elimination of chromatography and hazardous reagents translates directly into a more resilient and predictable supply chain, reducing the risk of batch failures and delivery delays.
- Cost Reduction in Manufacturing: The replacement of expensive and hazardous reagents like sodium hydride with commodity chemicals such as potassium hydroxide significantly lowers the raw material cost base. Furthermore, the removal of chromatographic purification steps eliminates the need for large volumes of high-purity organic solvents and silica gel, resulting in substantial savings in waste disposal and solvent recovery costs. The high isolated yields reported in the patent, such as 92% for the chloroalkyl pyridine and 94% for the final hydrogenation, ensure that material throughput is maximized, further driving down the cost per kilogram of the final intermediate.
- Enhanced Supply Chain Reliability: The use of robust, non-proprietary reagents ensures that the supply chain is not dependent on single-source vendors for exotic catalysts or ligands. Solvents like DMSO and acetic acid are widely available globally, mitigating the risk of regional shortages disrupting production schedules. Additionally, the simplified workup procedures, which rely on filtration and crystallization rather than complex distillations or extractions, reduce the processing time per batch. This efficiency allows for faster turnaround times and greater flexibility in responding to fluctuating market demand for fibrinogen antagonist APIs.
- Scalability and Environmental Compliance: From an environmental health and safety (EHS) perspective, avoiding sodium hydride removes a significant fire hazard from the manufacturing floor, simplifying facility permitting and insurance requirements. The aqueous workup conditions and the ability to recycle solvents like isopropyl acetate align well with green chemistry principles, reducing the overall environmental footprint of the manufacturing process. The process is inherently scalable, as demonstrated by the patent examples which describe operations in 50-liter reactors, providing a clear pathway for expansion to multi-ton commercial production without encountering unforeseen engineering hurdles.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthesis method. These insights are derived directly from the experimental data and process descriptions found within the patent documentation, providing clarity on critical control points.
Q: How does this synthesis method prevent racemization of the chiral center?
A: The process utilizes BSTFA for temporary bis-O,O'-silylation of L-tyrosine, which protects the chiral center during the subsequent N-sulfonylation step, thereby avoiding racemization and ensuring high optical purity.
Q: What are the advantages of using KOH/DMSO over traditional NaH/DMF for ether formation?
A: Using aqueous KOH in DMSO eliminates the safety hazards associated with sodium hydride (NaH) and avoids the difficult workup procedures required for DMF, while still achieving high yields of 85-96% for the ether coupling.
Q: How are dialkylated impurities removed without chromatography?
A: The method employs a precise pH adjustment strategy during crystallization. By adjusting the pH to the isoelectric point (pH 4.8) and subsequently washing with 10% acetic acid, dialkylated impurities and unreacted starting materials are effectively removed.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fibrinogen Receptor Antagonist Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a stable supply of high-quality intermediates for your antithrombotic drug development programs. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistency and precision. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of fibrinogen receptor antagonist intermediate meets the highest international standards for pharmaceutical use.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic advantages of switching to this methodology. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your supply chain goals.