Advanced Liquid Phase Synthesis of Buserelin: Scalable Technology for Global Pharmaceutical Manufacturing
Advanced Liquid Phase Synthesis of Buserelin: Scalable Technology for Global Pharmaceutical Manufacturing
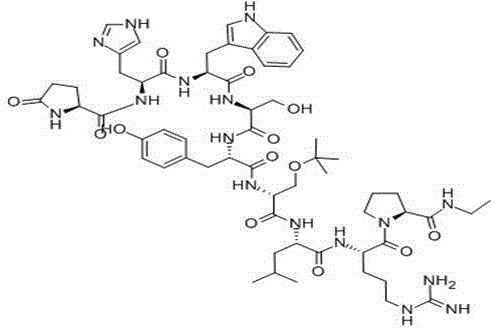
The pharmaceutical industry is constantly seeking more efficient pathways for the production of complex peptide therapeutics, and the liquid phase synthesis method of buserelin described in patent CN109438560B represents a significant technological leap forward. This innovative approach addresses the longstanding challenges associated with the manufacturing of Gonadotropin-Releasing Hormone (GnRH) analogues by shifting away from traditional, resource-intensive solid-phase strategies. By employing a sophisticated 5+4 fragment condensation technique, this method enables the direct coupling of a pentapeptide fragment and a tetrapeptide fragment under the influence of a specialized catalyst system. The core of this breakthrough lies in the utilization of 2-iodo-4-methoxy-phenylboronic acid combined with 4A molecular sieves, which creates a highly controlled reaction environment. This not only ensures exceptional product purity but also introduces a level of operational flexibility that is critical for modern supply chains. For R&D directors and procurement specialists alike, understanding this shift from linear stepwise synthesis to convergent fragment coupling is essential for evaluating the future cost structures and reliability of peptide API sourcing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional methodologies for synthesizing buserelin, particularly solid-phase peptide synthesis (SPPS), have long been plagued by inherent inefficiencies that impact both cost and quality at scale. In conventional solid-phase routes, the strategy often involves the selection of fully protected peptides followed by complex deprotection sequences using metal catalysts like palladium or harsh acidic conditions. A critical failure point in these legacy methods is the removal of side-chain protecting groups; for instance, the use of trifluoroacetic acid (TFA) to remove acid-sensitive groups can inadvertently cleave the protective groups on the Serine side chain, leading to the formation of difficult-to-remove impurities. Furthermore, the reliance on stoichiometric amounts of expensive coupling reagents for every single amino acid addition drives up the raw material costs exponentially as the peptide chain lengthens. The accumulation of deletion sequences and racemization byproducts during the lengthy cycle times of SPPS necessitates rigorous and costly purification steps, often resulting in lower overall yields that struggle to meet the demands of large-scale commercial production.
The Novel Approach
In stark contrast, the novel liquid-phase synthesis method outlined in the patent data revolutionizes the production landscape by adopting a convergent 5+4 fragment strategy. Instead of building the nonapeptide one amino acid at a time, this approach synthesizes two smaller, manageable fragments independently and then couples them in a single, highly efficient condensation step. This drastically reduces the total number of reaction cycles and exposure to potentially damaging conditions. The use of 2-iodo-4-methoxy-phenylboronic acid as a catalyst allows for the activation of the carboxyl terminal of the pentapeptide fragment under mild conditions, facilitating a clean reaction with the amino terminal of the tetrapeptide fragment. Crucially, this method avoids the harsh deprotection environments that compromise the Serine residue in traditional methods, thereby preserving the structural integrity of the final molecule. The ability to recycle the catalyst and the molecular sieves further enhances the economic viability of this process, making it a superior choice for manufacturers aiming to optimize their production workflows.
Mechanistic Insights into Boronic Acid-Catalyzed Fragment Condensation
The mechanistic elegance of this synthesis lies in the specific role of the 2-iodo-4-methoxy-phenylboronic acid catalyst, which acts as a potent Lewis acid to activate the carboxylic acid moiety of the pentapeptide fragment. In the presence of 4A molecular sieves, which serve as an effective water scavenger, the catalyst promotes the formation of a reactive acyl boronate intermediate or a mixed anhydride species in situ. This activation lowers the energy barrier for the nucleophilic attack by the free amine of the tetrapeptide fragment, driving the amide bond formation forward with high selectivity. The molecular sieves play a dual role: they sequester the water produced during the condensation reaction, shifting the equilibrium towards product formation according to Le Chatelier's principle, and they maintain an anhydrous environment that minimizes hydrolysis of the activated intermediates. This synergistic interaction between the boronic acid catalyst and the desiccant ensures that the reaction proceeds rapidly at room temperature, avoiding the thermal stress that can lead to epimerization or degradation of sensitive amino acid residues like Histidine and Tryptophan.
From an impurity control perspective, this mechanism offers distinct advantages over carbodiimide-based coupling methods often used in liquid phase synthesis. Traditional reagents like DCC can generate urea byproducts that are notoriously difficult to separate from the peptide product, whereas the boronic acid system generates cleaner reaction profiles. The specific protection strategy employed for the side chains—such as using Trt for Histidine and OtBu for Serine and Tyrosine—is carefully selected to be orthogonal to the condensation conditions. This ensures that the side chains remain intact during the fragment coupling, preventing the formation of branched peptides or deletion sequences that commonly arise from premature deprotection. The result is a crude product with significantly higher purity, which simplifies downstream processing and reduces the burden on chromatographic purification systems, ultimately leading to a more robust and reproducible manufacturing process.
How to Synthesize Buserelin Efficiently
The synthesis of buserelin via this advanced liquid-phase route involves a logical sequence of fragment preparation followed by the critical catalytic coupling step. The process begins with the independent assembly of the N-terminal pentapeptide fragment (Pyr-His-Trp-Ser-Tyr-OH) and the C-terminal tetrapeptide fragment (D-Ser-Leu-Arg-Pro-NHEt) using standard protection group chemistry. Once these high-purity building blocks are secured, they are brought together in the presence of the recyclable boronic acid catalyst and molecular sieves to form the full nonapeptide backbone. The detailed standardized synthetic steps, including specific solvent systems, molar ratios, and workup procedures required to achieve GMP-grade quality, are outlined in the technical guide below.
- Synthesize the protected pentapeptide fragment (Pyr-His-Trp-Ser-Tyr-OH) using stepwise activation and coupling.
- Prepare the protected tetrapeptide fragment (D-Ser-Leu-Arg-Pro-NHEt) through sequential amino acid addition.
- Condense the two fragments using 2-iodo-4-methoxy-phenylboronic acid catalyst and 4A molecular sieves, followed by deprotection.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this liquid-phase fragment condensation technology translates directly into tangible strategic benefits regarding cost stability and supply security. By eliminating the need for vast quantities of solid-phase resins and the associated swelling solvents, the process significantly reduces the volumetric footprint of the reaction, allowing for larger batch sizes in existing reactor infrastructure. The avoidance of stoichiometric coupling reagents for every single amino acid addition results in a drastic simplification of the bill of materials, reducing the exposure to price volatility of specialized peptide coupling agents. Furthermore, the recyclability of the catalyst system means that the effective cost per kilogram of the active catalyst is negligible over multiple batches, contributing to substantial long-term cost savings without compromising on reaction kinetics or yield.
- Cost Reduction in Manufacturing: The economic model of this synthesis is fundamentally superior because it decouples the production cost from the linear increase typically seen in peptide chain elongation. By condensing two large fragments rather than nine individual amino acids, the number of purification events is minimized, which is often the most expensive part of peptide manufacturing. The elimination of expensive heavy metal catalysts for deprotection, such as palladium, removes a significant cost center and eliminates the need for stringent residual metal testing and removal steps. This streamlined workflow ensures that the cost of goods sold (COGS) remains competitive even as production volumes scale up, providing a clear margin advantage for generic or biosimilar developers.
- Enhanced Supply Chain Reliability: Supply continuity is bolstered by the use of readily available starting materials and the robustness of the liquid-phase chemistry, which is less susceptible to the batch-to-batch variability often encountered with solid-phase resins. The ability to stockpile the two key peptide fragments independently allows for a flexible manufacturing schedule; if demand spikes, the final coupling step can be accelerated without needing to restart the entire synthesis from the first amino acid. This modularity reduces the lead time for high-purity buserelin batches, ensuring that pharmaceutical partners can maintain consistent inventory levels to meet market demand without the risk of prolonged production bottlenecks.
- Scalability and Environmental Compliance: From an environmental and regulatory standpoint, this method aligns perfectly with green chemistry principles by reducing solvent consumption and chemical waste generation. The liquid-phase process avoids the massive volumes of DMF and DCM typically required for washing solid-phase resins, simplifying waste treatment and lowering disposal costs. The mild reaction conditions and the absence of toxic heavy metals in the final deprotection steps facilitate easier regulatory approval and compliance with increasingly strict environmental standards. This scalability ensures that the process can be seamlessly transferred from pilot plant operations to multi-ton commercial production without the need for fundamental re-engineering of the process parameters.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this liquid-phase synthesis technology. These answers are derived directly from the patent specifications and are intended to clarify the operational benefits and chemical rationale behind the 5+4 fragment condensation strategy. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this method for their own manufacturing portfolios.
Q: What are the primary advantages of this liquid-phase method over traditional solid-phase synthesis?
A: This method utilizes a 5+4 fragment condensation strategy which significantly reduces the number of reaction cycles compared to stepwise solid-phase synthesis. It avoids the use of harsh deprotection conditions that often lead to side-chain impurities, specifically preserving the integrity of the Serine residue, and allows for the recycling of the boronic acid catalyst.
Q: How does the 2-iodo-4-methoxy-phenylboronic acid catalyst improve the process?
A: The catalyst facilitates the direct condensation of the pentapeptide and tetrapeptide fragments under mild conditions. It activates the carboxyl group efficiently without requiring excessive amounts of traditional coupling reagents, thereby reducing chemical waste and simplifying the purification process while maintaining high stereochemical integrity.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the process is specifically designed for industrial scalability. By avoiding the limitations of resin loading capacity inherent in solid-phase synthesis and utilizing recoverable catalysts and molecular sieves, the method offers a robust pathway for manufacturing multi-kilogram to ton-scale quantities of high-purity Buserelin.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Buserelin Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of complex peptides like buserelin requires more than just a patent; it demands deep process engineering expertise and a commitment to quality. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. Our facilities are equipped with state-of-the-art reactors capable of handling the specific solvent and temperature requirements of boronic acid-catalyzed reactions, and our rigorous QC labs enforce stringent purity specifications to guarantee that every batch meets the highest international pharmacopoeia standards.
We invite you to collaborate with us to leverage this advanced synthesis technology for your next project. Our technical team is ready to provide a Customized Cost-Saving Analysis that demonstrates how switching to this liquid-phase method can optimize your budget. Please contact our technical procurement team today to request specific COA data and route feasibility assessments tailored to your volume requirements, and let us help you secure a stable, high-quality supply of buserelin for the global market.