Scalable Preparation of Buserelin and Goserelin via Selective Hydrogenolysis and Solid-Phase Synthesis
Scalable Preparation of Buserelin and Goserelin via Selective Hydrogenolysis and Solid-Phase Synthesis
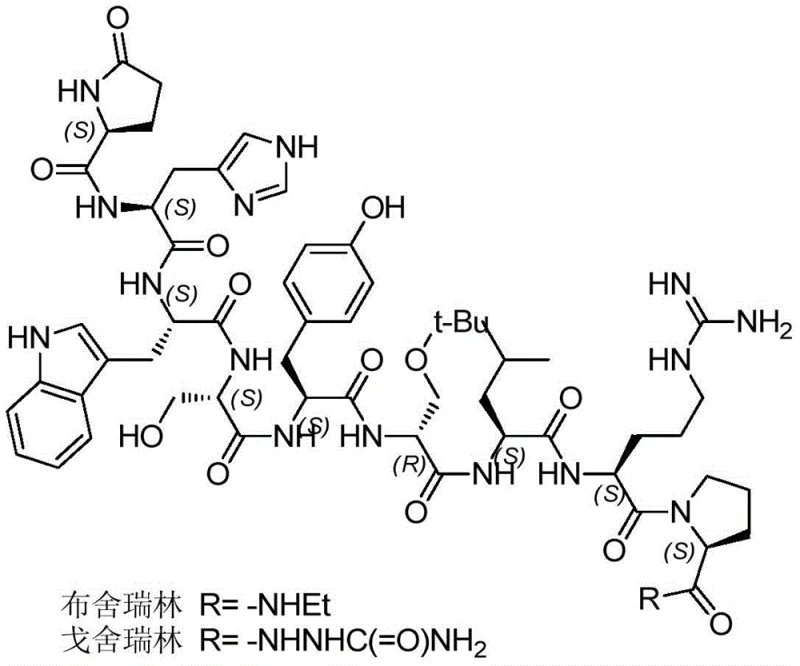
The pharmaceutical industry continuously seeks robust manufacturing routes for gonadotropin-releasing hormone (GnRH) analogues, specifically Buserelin and Goserelin, which are critical in the treatment of prostate cancer and endometriosis. A recent technological breakthrough, detailed in patent CN107540727B, introduces a highly efficient preparation method that addresses long-standing challenges in peptide deprotection and solubility. This novel approach leverages a modified solid-phase peptide synthesis (SPPS) strategy combined with a unique catalytic hydrogenolysis system utilizing pyridine hydrochloride or acetic acid aqueous solutions. By optimizing the solvent environment for the final deprotection step, this method achieves superior selectivity, preserving acid-sensitive protecting groups while ensuring high crude purity. For global procurement teams and R&D directors, this represents a significant advancement in the reliable supply of high-purity pharmaceutical intermediates, offering a pathway to reduce production costs and enhance process stability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Buserelin and Goserelin has been plagued by the incompatibility of protecting group strategies with industrial scalability. Conventional liquid-phase synthesis is operationally complex, requiring purification after every step, which renders it economically unviable for large-scale production. While solid-phase synthesis is preferred, standard Fmoc/tBu strategies face a critical bottleneck: the D-Ser(tBu) residue is highly sensitive to acidic conditions. Traditional deprotection methods often employ high concentrations of trifluoroacetic acid (TFA) in dichloromethane to remove trityl (Trt) groups from Histidine. However, these harsh acidic conditions frequently cause the premature cleavage of the t-butyl ether on the D-Ser side chain, generating difficult-to-remove impurities that compromise the final API quality. Furthermore, existing hydrogenolysis methods for removing benzyl (Bzl) and nitro (NO2) groups suffer from poor solubility of the fully protected peptide in standard solvents like methanol or ethanol, leading to slow reaction rates and the need for excessive catalyst loading, which introduces safety hazards and filtration difficulties.
The Novel Approach
The methodology disclosed in patent CN107540727B fundamentally reengineers the deprotection workflow to overcome these solubility and selectivity barriers. Instead of relying on aggressive acidolysis or inefficient standard hydrogenation, this process utilizes a specialized solvent system designated as 'Solvent Z'. This solvent is either a methanol solution containing 5% pyridine hydrochloride or an aqueous acetic acid solution with a concentration of 85-95%. This innovation dramatically improves the solubility of the fully protected peptide intermediates, allowing for homogeneous catalytic hydrogenolysis using palladium on carbon (Pd/C) at moderate temperatures (58-65°C). The result is a highly selective removal of the benzyl and nitro protecting groups without affecting the critical t-butyl ether on the D-Ser residue. This single-step deprotection replaces the multi-step, hazardous protocols of the past, streamlining the transition from fully protected peptide to the final active pharmaceutical ingredient with exceptional efficiency.
Mechanistic Insights into Selective Catalytic Hydrogenolysis
The core chemical innovation lies in the modulation of the reaction medium during the palladium-catalyzed hydrogenolysis step. In traditional settings, the low solubility of the protected nonapeptide in alcohols creates a heterogeneous mixture where mass transfer limits the reaction rate, often requiring prolonged exposure to hydrogen and catalyst. By introducing pyridine hydrochloride or high-concentration acetic acid, the protonation state of the peptide backbone and side chains is altered, significantly enhancing solvation in the polar organic medium. This ensures that the Pd/C catalyst has uniform access to the benzyl and nitro moieties located on the Tyrosine, Serine, and Arginine residues. The mechanism proceeds via the adsorption of hydrogen onto the palladium surface, followed by the reductive cleavage of the C-O and C-N bonds of the protecting groups. Crucially, the mild acidity of the pyridine hydrochloride or dilute acetic acid system is insufficient to protonate and cleave the sterically hindered t-butyl ether linkage on the D-Ser side chain, thereby preventing the formation of des-t-butyl impurities that plague TFA-based methods.
Impurity control is further enhanced by the initial mild cleavage from the 2-CTC resin. By using a very low concentration of TFA (1-3%) in dichloromethane, the peptide is released from the solid support as a fully protected species with all side-chain protecting groups intact. This contrasts sharply with high-acid cleavage cocktails that might partially deprotect sensitive residues prematurely. The subsequent coupling of the C-terminal tail—ethylamine for Buserelin or semicarbazide for Goserelin—is performed in solution phase using EDCI/NMM activation, ensuring high conversion before the final global deprotection. This sequence minimizes the accumulation of deletion sequences or truncated peptides, resulting in a crude product with HPLC purity often exceeding 90% before final preparative chromatography, which significantly reduces the load on downstream purification units.

How to Synthesize Buserelin and Goserelin Efficiently
The synthesis protocol outlined in the patent provides a clear, scalable roadmap for manufacturing these complex hormones. The process begins with the loading of Fmoc-Proline onto 2-chlorotrityl chloride (2-CTC) resin, followed by the sequential addition of amino acids using standard Fmoc chemistry with DIPCDI/HOAt or HATU activation. Once the linear nonapeptide chain is assembled, it is cleaved under exceptionally mild acidic conditions to preserve the integrity of the protecting groups. The free C-terminus is then activated and coupled with the appropriate amine derivative to form the fully protected precursor. The final and most critical transformation involves the hydrogenolysis in the specialized solvent system, which simultaneously removes the remaining protecting groups to yield the bioactive peptide. For detailed operational parameters, stoichiometry, and specific workup procedures, please refer to the standardized synthesis guide below.
- Load Fmoc-Pro-OH onto 2-CTC resin and perform Fmoc-SPPS to assemble the fully protected nonapeptide resin.
- Cleave the peptide from the resin using mild 1-3% TFA/DCM to obtain the fully protected free peptide without removing acid-sensitive groups.
- Couple the C-terminal tail (ethylamine or semicarbazide) and perform catalytic hydrogenolysis using Pd/C in 5% pyridine hydrochloride/methanol or 85-95% acetic acid aqueous solution.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers tangible strategic benefits beyond mere technical elegance. The primary advantage lies in the drastic simplification of the deprotection workflow. By eliminating the need for multi-step deprotection sequences involving harsh acids and intermediate isolations, the overall processing time is significantly reduced. This streamlining directly translates to lower labor costs and reduced consumption of expensive reagents and solvents. Furthermore, the enhanced solubility of the intermediates in the pyridine/acetic acid system allows for higher concentration reactions, which increases the throughput of existing reactor vessels without the need for capital expenditure on larger equipment. This efficiency gain is critical for maintaining competitive pricing in the global market for GnRH analogues.
- Cost Reduction in Manufacturing: The elimination of complex, multi-stage deprotection protocols significantly lowers the operational expenditure associated with reagent consumption and waste disposal. By avoiding the use of large volumes of dichloromethane for repeated TFA treatments and reducing the requirement for excessive palladium catalyst due to improved solubility, the direct material costs are substantially optimized. Additionally, the higher crude purity achieved through this selective method reduces the burden on preparative HPLC purification, extending column life and decreasing the volume of mobile phase required, which represents a major cost center in peptide manufacturing.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route ensures consistent batch-to-batch quality, which is paramount for maintaining regulatory compliance and avoiding costly production delays. The use of stable, commercially available reagents like pyridine hydrochloride and acetic acid mitigates the risk of supply disruptions often associated with specialized or hazardous deprotection agents. Moreover, the mild reaction conditions reduce the risk of equipment corrosion and safety incidents, ensuring uninterrupted production schedules and reliable delivery timelines for downstream pharmaceutical customers.
- Scalability and Environmental Compliance: This process is inherently designed for scale-up, moving away from laboratory-centric techniques to industrial-friendly operations. The ability to perform the final deprotection in a single pot with high selectivity minimizes the generation of hazardous waste streams associated with multiple workup and purification steps. The reduced reliance on chlorinated solvents for deprotection aligns with increasingly stringent environmental regulations, facilitating easier permitting and sustainability reporting for manufacturing sites aiming to reduce their carbon footprint and chemical waste output.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation of this synthesis method. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, focusing on the practical implications for process chemistry and quality control. Understanding these nuances is essential for technical teams evaluating the feasibility of technology transfer.
Q: How does the new solvent system improve the deprotection of Buserelin compared to traditional methods?
A: Traditional methods often rely on strong acids like TFA which can inadvertently remove the acid-sensitive t-butyl group on the D-Ser residue, leading to significant impurities. The patented method utilizes a specific solvent system (5% pyridine hydrochloride in methanol or 85-95% acetic acid aqueous solution) during palladium-catalyzed hydrogenolysis. This environment allows for the selective removal of benzyl and nitro protecting groups while preserving the t-butyl ether, resulting in crude peptide purity exceeding 90% prior to final purification.
Q: What are the solubility advantages of using pyridine hydrochloride or acetic acid in the hydrogenolysis step?
A: Fully protected Buserelin and Goserelin peptides exhibit very poor solubility in standard hydrogenolysis solvents like pure methanol or ethanol, which necessitates large solvent volumes or excessive catalyst loading. By employing a methanol solution of 5% pyridine hydrochloride or an 85-95% acetic acid aqueous solution, the solubility of the fully protected intermediates is drastically improved. This ensures homogeneous reaction conditions, faster reaction kinetics, and eliminates safety hazards associated with heterogeneous slurry reactions.
Q: Is this synthesis route suitable for large-scale commercial production?
A: Yes, the process is specifically designed for industrial scalability. It avoids complex multi-step deprotection sequences and utilizes robust solid-phase synthesis on 2-CTC resin followed by solution-phase tail coupling. The mild cleavage conditions (1-3% TFA) and the efficient single-step hydrogenolysis reduce operational complexity and equipment corrosion risks, making it highly viable for manufacturing at the 100 kg to multi-ton scale.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Buserelin and Goserelin Supplier
As the demand for high-quality GnRH analogues continues to grow, partnering with a manufacturer that possesses deep technical expertise in peptide synthesis is essential. NINGBO INNO PHARMCHEM stands at the forefront of this sector, leveraging advanced methodologies like the one described in CN107540727B to deliver superior products. Our facility is equipped with extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous volume requirements of global pharmaceutical contracts. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Buserelin or Goserelin meets the highest international pharmacopoeia standards, providing our partners with absolute confidence in their supply chain.
We invite you to engage with our technical team to explore how this optimized synthesis route can benefit your specific project needs. Whether you require custom synthesis services or bulk supply of these critical intermediates, we are prepared to provide a Customized Cost-Saving Analysis tailored to your production targets. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our commitment to innovation can drive value for your organization.