Advanced Synthesis of Florfenicol via Aziridine Fluorination for Commercial Scale Veterinary API Production
The pharmaceutical landscape for veterinary antibiotics is constantly evolving, with a persistent demand for more efficient and scalable synthesis routes for broad-spectrum agents like Florfenicol. Patent CN102070497A introduces a significant technological advancement in this domain, detailing a novel synthetic methodology that addresses the longstanding inefficiencies associated with traditional chloramphenicol analog production. This patent outlines a streamlined four-step sequence starting from a chiral aziridine intermediate, leveraging specific fluorination strategies to install the critical fluoromethyl group with high stereochemical fidelity. By circumventing the need for complex resolution steps often found in legacy processes, this invention offers a robust pathway that aligns perfectly with modern green chemistry principles and industrial scalability requirements. For R&D directors and procurement specialists alike, understanding the nuances of this aziridine-based approach is crucial for optimizing supply chains and reducing the cost of goods sold for high-volume veterinary APIs.
The Limitations of Conventional Methods vs. The Novel Approach
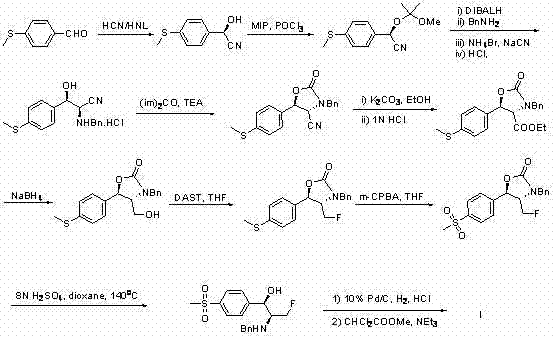
The Limitations of Conventional Methods
Historically, the synthesis of Florfenicol has been plagued by cumbersome multi-step sequences that rely heavily on the resolution of racemic mixtures or the use of hazardous reagents in excessive quantities. As illustrated in prior art such as Chinese Patent CN1743308, traditional routes often involve asymmetric cyanohydrin formation followed by a myriad of protection and deprotection cycles, leading to suboptimal overall yields and significant environmental burdens. These conventional pathways typically necessitate the construction of chiral centers through induction methods that require stringent temperature controls and expensive catalysts, thereby inflating the operational expenditure for manufacturers. Furthermore, the reliance on starting materials derived from splitting processes results in the discard of unwanted enantiomers, representing a massive loss of raw material value and creating substantial waste disposal challenges for chemical plants. The complexity of these older routes also introduces multiple opportunities for impurity generation, complicating downstream purification and quality control efforts.
The Novel Approach
In stark contrast to the convoluted legacy methods, the methodology described in CN102070497A utilizes a strategic aziridine intermediate to achieve the target molecular architecture with remarkable efficiency. This innovative route capitalizes on the inherent ring strain of the aziridine moiety to facilitate regioselective ring opening, thereby establishing the necessary carbon-nitrogen and carbon-oxygen bonds in a single transformative step. By employing readily available fluorinating agents such as DAST or Ishikawa reagent on a protected aziridine alcohol, the process installs the vital fluoromethyl group early in the sequence, ensuring that subsequent transformations proceed with high chemoselectivity. The elimination of resolution steps is a pivotal improvement, as it allows for the theoretical utilization of all starting material mass, drastically improving the atom economy of the entire process. This streamlined approach not only shortens the production timeline but also simplifies the equipment requirements, making it highly attractive for large-scale commercial manufacturing where throughput and consistency are paramount.
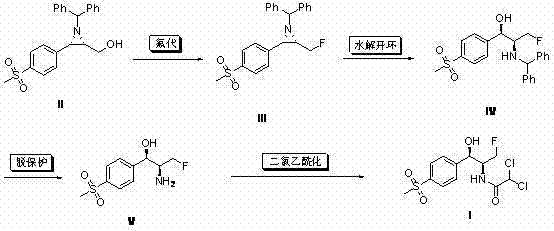
Mechanistic Insights into Aziridine Fluorination and Ring Opening
The core of this synthetic breakthrough lies in the precise execution of the nucleophilic fluorination step, where a hydroxymethyl aziridine is converted into a fluoromethyl aziridine using reagents like DAST or BAST. Mechanistically, this transformation involves the activation of the hydroxyl group by the sulfur species, followed by a nucleophilic attack by fluoride ion, proceeding with inversion of configuration if the carbon center is chiral, although in this specific primary alcohol context, it serves primarily to swap the leaving group. The choice of fluorinating agent is critical; while DAST is highly reactive, the patent also validates the use of Ishikawa reagent at elevated temperatures, offering flexibility for facilities that may prioritize thermal stability over cryogenic handling. Following fluorination, the acid-catalyzed ring opening of the aziridine ring is a masterclass in regiocontrol, where the protonated nitrogen acts as a leaving group facilitator, allowing a nucleophile (typically water or solvent) to attack the benzylic position. This step effectively unlocks the three-membered ring to reveal the 1,2-amino alcohol scaffold essential for Florfenicol's biological activity, all while maintaining the stereochemical integrity established in the precursor.
Impurity control in this process is inherently managed through the high selectivity of the ring-opening reaction and the subsequent hydrogenolysis step. The use of p-toluenesulfonic acid for ring opening ensures that side reactions such as polymerization or elimination are minimized, provided the stoichiometry and temperature are kept within the specified ranges of 0°C to 80°C. Furthermore, the final deprotection via palladium-carbon hydrogenation is a clean method for removing the benzhydryl protecting group, yielding the free amine without introducing halogenated impurities that might arise from acidic hydrolysis of amides. The final dichloroacetylation is performed under mild basic conditions using triethylamine or similar amines, which scavenges the generated acid and drives the equilibrium towards the desired amide product. This careful orchestration of reaction conditions ensures that the final API meets stringent purity specifications, reducing the burden on crystallization and chromatographic purification stages.
How to Synthesize Florfenicol Efficiently
The synthesis of Florfenicol via this patented route involves a logical progression of functional group manipulations that prioritize yield and safety. The process begins with the fluorination of the aziridine alcohol, followed by acid-mediated ring opening to generate the amino alcohol backbone. Subsequent removal of the protecting group via hydrogenation reveals the primary amine, which is finally acylated to complete the molecule. For detailed operational parameters, including specific solvent ratios, quenching procedures, and workup protocols, please refer to the standardized synthesis guide below which encapsulates the critical process parameters defined in the patent documentation.
- Perform nucleophilic fluorination on a chiral aziridine alcohol intermediate using DAST, BAST, or Ishikawa reagent to generate the fluoromethyl aziridine derivative.
- Execute acid-catalyzed ring opening of the fluorinated aziridine using p-toluenesulfonic acid to establish the requisite amino alcohol backbone.
- Conduct palladium-carbon hydrogenation for deprotection followed by dichloroacetylation under basic conditions to finalize the Florfenicol active pharmaceutical ingredient.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this aziridine-based synthesis route offers profound advantages in terms of cost structure and logistical reliability. By eliminating the need for chiral resolution of starting materials, manufacturers can significantly reduce the volume of raw materials required per kilogram of finished product, directly translating to lower input costs and reduced waste disposal fees. The use of common fluorinating reagents and standard acid catalysts means that the supply chain is not dependent on exotic or single-source specialty chemicals, thereby mitigating the risk of supply disruptions that often plague complex pharmaceutical syntheses. Additionally, the mild reaction conditions, particularly the ability to perform ring opening at near-ambient temperatures, reduce the energy consumption associated with heating and cooling large-scale reactors, contributing to a lower carbon footprint and operational expenditure. The robustness of the process also implies higher batch success rates, ensuring consistent delivery schedules for downstream formulators who rely on a steady stream of high-quality veterinary API intermediates.
- Cost Reduction in Manufacturing: The elimination of resolution steps and the reduction in total synthetic steps lead to a substantial decrease in labor, solvent, and utility costs per unit of production. By avoiding the discard of unwanted enantiomers, the effective yield from raw material to final product is maximized, providing a clear economic advantage over legacy methods that suffer from inherent 50% theoretical yield losses during resolution. Furthermore, the use of catalytic hydrogenation for deprotection avoids the generation of stoichiometric salt waste associated with chemical deprotection methods, simplifying effluent treatment and lowering environmental compliance costs.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as p-toluenesulfonic acid, triethylamine, and standard fluorinating agents ensures that the manufacturing process is resilient to market fluctuations in specialty reagent pricing. The simplified workflow reduces the number of intermediate isolation points, which minimizes the risk of material degradation during storage and transport between production stages. This continuity enhances the predictability of lead times, allowing supply chain managers to maintain leaner inventory levels while still meeting the rigorous demand schedules of the global veterinary pharmaceutical market.
- Scalability and Environmental Compliance: The process is designed with industrial scalability in mind, utilizing reaction conditions that are easily transferable from pilot plant to multi-ton commercial production without significant re-engineering. The avoidance of highly toxic cyanide reagents, which are prevalent in other reported syntheses, drastically improves the safety profile of the manufacturing facility and simplifies regulatory approval processes. The overall reduction in solvent usage and waste generation aligns with increasingly strict global environmental regulations, future-proofing the production asset against tightening ecological standards and potential carbon taxes.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Florfenicol synthesis technology. These answers are derived directly from the experimental data and claims presented in patent CN102070497A, providing a reliable foundation for feasibility assessments and technology transfer discussions. Understanding these specifics is vital for engineering teams evaluating the retrofitting of existing production lines or the commissioning of new dedicated suites for veterinary antibiotic manufacture.
Q: What fluorinating agents are compatible with this aziridine route?
A: The patent specifies the use of DAST, BAST, or Ishikawa reagent, allowing flexibility in reagent sourcing based on availability and safety protocols.
Q: How does this method improve upon traditional thiamphenicol derivatization?
A: Unlike methods requiring resolution of racemic mixtures, this route utilizes chiral aziridine precursors, significantly reducing waste and improving overall atom economy.
Q: What are the critical reaction conditions for the ring-opening step?
A: The ring opening proceeds efficiently using p-toluenesulfonic acid in solvents like acetonitrile or dichloromethane at moderate temperatures ranging from 0°C to 80°C.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Florfenicol Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and compliant synthesis routes in the competitive veterinary pharmaceutical market. Our team of expert process chemists has extensively analyzed the technology disclosed in CN102070497A and possesses the capability to implement this advanced aziridine-based methodology at commercial scale. We boast extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with unwavering consistency. Our state-of-the-art facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of Florfenicol intermediate or API meets the highest international standards for safety and efficacy.
We invite you to collaborate with us to leverage this cutting-edge synthesis technology for your product portfolio. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this route can optimize your bottom line. Please contact us today to request specific COA data and route feasibility assessments, and let us partner with you to drive innovation and efficiency in your veterinary drug supply chain.