Scalable Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Advanced Pharmaceutical Intermediates
Scalable Synthesis of 5-Trifluoromethyl-1,2,4-Triazoles for Advanced Pharmaceutical Intermediates
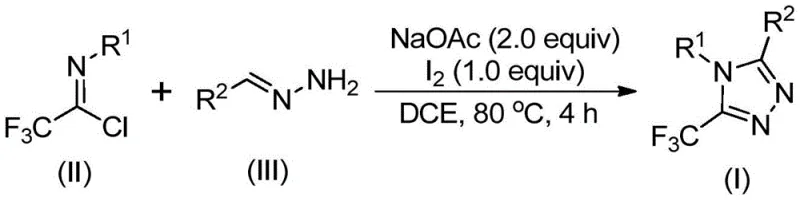
The pharmaceutical and fine chemical industries are constantly seeking robust methodologies to construct nitrogen-containing heterocycles, particularly those incorporating fluorine motifs which are pivotal for enhancing metabolic stability and bioavailability. Patent CN110467579B discloses a groundbreaking preparation method for 5-trifluoromethyl substituted 1,2,4-triazole compounds, addressing critical bottlenecks in current synthetic routes. This technology leverages a non-metallic iodine-promoted cyclization strategy that transforms inexpensive hydrazones and trifluoroethylimidoyl chlorides into high-value scaffolds under mild conditions. The significance of this development cannot be overstated, as 1,2,4-triazole cores are ubiquitous in modern medicinal chemistry, serving as key structural elements in antifungal agents, kinase inhibitors, and functional materials. By eliminating the reliance on hazardous trifluoromethylating agents and complex catalytic systems, this invention offers a streamlined pathway for the production of reliable pharmaceutical intermediate supplier candidates.
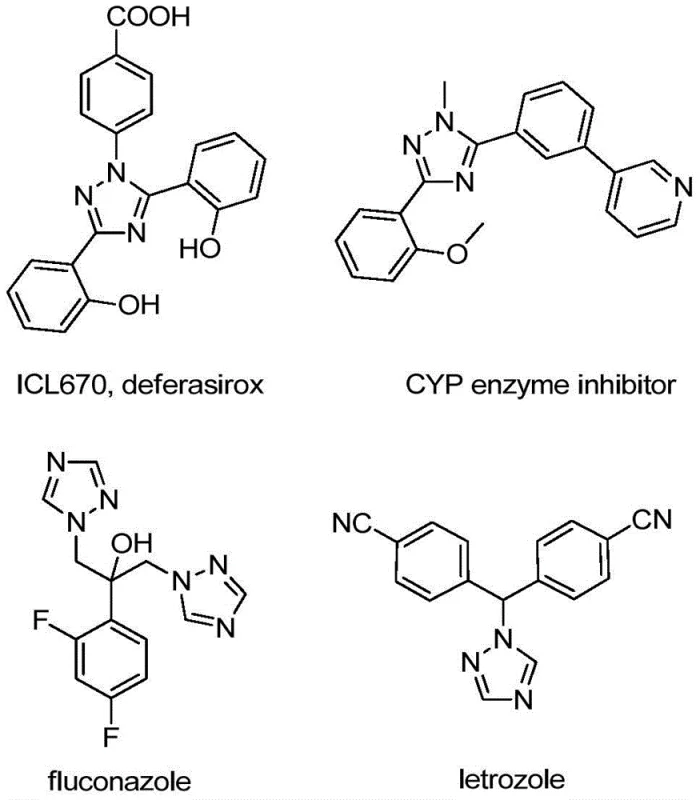
The strategic importance of the 1,2,4-triazole ring system is evident when examining the landscape of approved drugs and clinical candidates. As illustrated in the provided chemical structures, molecules like deferasirox, fluconazole, and letrozole rely on this heterocyclic framework to achieve their therapeutic efficacy. The introduction of a trifluoromethyl group at the 5-position further amplifies the pharmacological potential by modulating lipophilicity and electronic properties, which are essential parameters for optimizing drug-receptor interactions. Consequently, developing a synthesis method that can access these diversified structures with high efficiency and low environmental impact is a priority for any organization focused on cost reduction in API manufacturing. The patented process described herein not only meets these criteria but also provides a versatile platform for generating libraries of novel compounds for drug discovery programs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of trifluoromethyl-substituted nitrogen heterocycles has been fraught with significant technical and economic challenges that hinder large-scale adoption. Traditional literature reports primarily describe two approaches: the direct trifluoromethylation of pre-synthesized heterocycles or the cycloaddition of trifluoromethyl synthons with coupling substrates. The former often necessitates the use of specialized and expensive trifluoromethylating reagents that can be difficult to handle and store safely. The latter frequently relies on unstable precursors such as trifluorodiazoethane, which poses severe safety risks due to its explosive nature and requires stringent safety protocols that inflate operational costs. Furthermore, many existing methods depend on transition metal catalysts that introduce the risk of heavy metal contamination in the final product, necessitating costly and time-consuming purification steps to meet regulatory standards for pharmaceutical ingredients. These limitations collectively create a barrier to entry for the commercial scale-up of complex polymer additives or pharmaceutical intermediates requiring this specific motif.
The Novel Approach
In stark contrast to these cumbersome legacy techniques, the novel approach detailed in patent CN110467579B utilizes a simple yet highly effective iodine-promoted cyclization strategy. This method employs readily available starting materials, specifically trifluoroethylimidoyl chloride and various hydrazones, which are derived from cheap aldehydes and hydrazine hydrate. The reaction proceeds in common organic solvents like dichloroethane at moderate temperatures ranging from 80°C to 100°C, completely bypassing the need for anhydrous or anaerobic conditions. This operational simplicity is a game-changer for process chemistry, as it allows reactions to be conducted in standard glassware without the need for specialized inert atmosphere equipment. The use of elemental iodine as a promoter rather than a stoichiometric reagent or toxic heavy metal catalyst ensures that the process remains environmentally benign while maintaining high conversion rates. This shift towards safer, more accessible reagents represents a significant advancement in the field of heterocyclic synthesis.
Mechanistic Insights into Iodine-Promoted Oxidative Cyclization
A deep understanding of the reaction mechanism is crucial for R&D teams aiming to optimize this process for specific substrates. The transformation likely initiates with a base-promoted intermolecular carbon-nitrogen bond formation between the trifluoroethylimidoyl chloride and the hydrazone, yielding a trifluoroacetamidine intermediate. This species subsequently undergoes isomerization to align the reactive centers for cyclization. The addition of elemental iodine then triggers a base-promoted oxidative iodination, generating a reactive iodine-containing intermediate. This key step facilitates an intramolecular electrophilic substitution reaction, where the nitrogen nucleophile attacks the activated carbon center, closing the five-membered ring. The final stage involves aromatization, driven by the elimination of hydrogen iodide, to furnish the stable 5-trifluoromethyl substituted 1,2,4-triazole product. This mechanistic pathway highlights the dual role of the reaction conditions in both building the molecular skeleton and establishing the aromatic character of the heterocycle.
From an impurity control perspective, the mildness of this oxidative cyclization offers distinct advantages over harsher acidic or basic conditions often found in alternative routes. The use of sodium acetate as a mild base helps to buffer the reaction environment, minimizing the formation of degradation byproducts that could arise from the hydrolysis of the imidoyl chloride or the hydrazone. Furthermore, the specificity of the iodine-mediated oxidation reduces the likelihood of over-oxidation or non-selective halogenation of the aromatic rings attached to the R1 and R2 positions. The patent data indicates that a wide range of substituents, including electron-donating groups like methyl and methoxy, as well as electron-withdrawing groups like bromo and nitro, are well-tolerated. This broad functional group compatibility ensures that the impurity profile remains manageable even when synthesizing complex derivatives, thereby simplifying the downstream purification workflow and enhancing the overall yield of high-purity OLED material or API precursors.
How to Synthesize 5-Trifluoromethyl-1,2,4-Triazole Efficiently
The execution of this synthesis protocol is designed to be straightforward, enabling rapid translation from laboratory bench to pilot plant. The process begins by combining the trifluoroethylimidoyl chloride, the selected hydrazone derivative, and sodium acetate in a suitable organic solvent such as dichloroethane. The mixture is heated to approximately 80°C and stirred for an initial period of 2 to 4 hours to allow for the formation of the amidine intermediate. Following this incubation, elemental iodine is introduced to the reaction vessel to drive the oxidative cyclization to completion over an additional 1 to 2 hours. Upon confirmation of reaction completion via standard analytical techniques, the mixture is subjected to a simple workup involving filtration and silica gel treatment, followed by column chromatography to isolate the pure product. For a detailed breakdown of the specific molar ratios and temperature profiles optimized for different substrates, please refer to the standardized synthesis steps outlined below.
- Mix sodium acetate, trifluoroethylimidoyl chloride, and hydrazone in an organic solvent such as dichloroethane.
- Heat the mixture to 80-100°C and react for 2-4 hours to facilitate initial bond formation.
- Add elemental iodine to the system and continue reacting for 1-2 hours to complete the oxidative cyclization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers compelling economic and logistical benefits that extend beyond mere chemical yield. The primary driver for cost optimization lies in the selection of raw materials; hydrazones are easily synthesized from abundant aldehydes and hydrazine, while trifluoroethylimidoyl chlorides are accessible commodity chemicals. This reliance on bulk-available feedstocks mitigates the risk of supply chain disruptions that often plague processes dependent on exotic or custom-synthesized reagents. Moreover, the elimination of heavy metal catalysts removes the necessity for expensive scavenging resins and extensive metal testing, which are significant cost centers in the manufacture of high-purity pharmaceutical intermediates. The simplified operational requirements, specifically the lack of need for inert gas lines or gloveboxes, further reduce capital expenditure and utility costs associated with facility maintenance.
- Cost Reduction in Manufacturing: The economic viability of this process is significantly enhanced by the avoidance of precious metal catalysts and hazardous reagents. By utilizing elemental iodine and sodium acetate, which are inexpensive and widely available, the direct material costs are drastically lowered compared to palladium or copper-catalyzed alternatives. Additionally, the simplified post-treatment procedure, which avoids complex aqueous workups or distillation steps required to remove volatile toxic reagents, leads to substantial savings in labor and waste disposal expenses. The high atom economy of the cyclization step ensures that a greater proportion of the input mass is converted into valuable product, minimizing the financial loss associated with unreacted starting materials.
- Enhanced Supply Chain Reliability: Stability of supply is a critical metric for any long-term manufacturing partnership, and this method excels by utilizing robust and shelf-stable starting materials. Unlike trifluorodiazoethane, which requires on-site generation and immediate consumption due to its instability, the reagents used in this protocol can be stored for extended periods without degradation. This flexibility allows manufacturers to maintain strategic stockpiles of raw materials, buffering against market volatility and ensuring continuous production schedules. The tolerance for ambient atmospheric conditions also means that the reaction is less susceptible to failures caused by minor leaks in reactor seals, further guaranteeing consistent batch-to-batch output and reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: Scaling chemical processes often introduces new safety and environmental challenges, but this iodine-promoted method is inherently designed for expansion. The reaction exotherm is manageable, and the absence of explosive intermediates makes it suitable for large-scale reactors without requiring specialized explosion-proof infrastructure. From an environmental standpoint, the process generates minimal hazardous waste, as the byproducts are primarily inorganic salts and recoverable solvents. This alignment with green chemistry principles facilitates easier regulatory approval and reduces the burden of environmental compliance reporting, making it an attractive option for companies aiming to reduce their carbon footprint while maintaining high production volumes.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of this technology for their specific applications, we have compiled a set of frequently asked questions based on the experimental data and technical specifications provided in the patent documentation. These inquiries address common concerns regarding reaction conditions, substrate scope, and purification requirements. Understanding these nuances is essential for integrating this methodology into existing production workflows or research pipelines. The answers provided below are derived directly from the validated results of the invention, ensuring accuracy and reliability for your decision-making process.
Q: What are the advantages of this iodine-promoted method over traditional trifluoromethylation?
A: This method avoids the use of dangerous reagents like trifluorodiazoethane and eliminates the need for expensive heavy metal catalysts, significantly simplifying purification and reducing safety risks.
Q: Does this synthesis require strict anhydrous or anaerobic conditions?
A: No, one of the key operational benefits of this patented process is that it proceeds efficiently under standard atmospheric conditions without the need for rigorous exclusion of moisture or oxygen.
Q: What is the substrate scope for R1 and R2 groups in this reaction?
A: The method demonstrates broad tolerance, accommodating substituted or unsubstituted aryl groups, alkenyl groups, and heteroaryl groups, allowing for the design of diverse molecular scaffolds.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Trifluoromethyl-1,2,4-Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced synthetic methodologies like the iodine-promoted cyclization described in patent CN110467579B. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent and reliable supply of critical intermediates. Our state-of-the-art facilities are equipped to handle the specific requirements of halogenated heterocycle synthesis, adhering to stringent purity specifications and rigorous QC labs to guarantee that every batch meets the highest industry standards. We are committed to leveraging our technical expertise to optimize this route for your specific needs, delivering cost-effective solutions without compromising on quality or safety.
We invite you to explore how our capabilities can support your drug development or material science projects. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments for your target molecules. Let us help you navigate the complexities of chemical manufacturing and accelerate your path to market with confidence and efficiency.