Advanced Synthesis of Amino Acid Intermediates: A Cost-Effective Negishi Coupling Strategy for Commercial Scale-Up
Advanced Synthesis of Amino Acid Intermediates: A Cost-Effective Negishi Coupling Strategy for Commercial Scale-Up
The pharmaceutical industry constantly seeks more efficient pathways to access complex chiral building blocks, particularly for the synthesis of potent small molecule inhibitors. Patent CN111471003B introduces a groundbreaking preparation method for a specific class of amino acid compounds, designated generally as Formula IV, which serve as critical intermediates in the manufacture of integrin inhibitors and other therapeutic agents. This technology represents a significant leap forward in process chemistry, addressing long-standing challenges related to cost, step economy, and raw material availability. By leveraging a palladium-catalyzed Negishi coupling strategy, the inventors have devised a route that bypasses the need for expensive chiral pool starting materials, instead constructing the carbon-carbon bond with high stereochemical fidelity using readily available precursors. For R&D directors and procurement specialists alike, this patent offers a compelling alternative to legacy synthesis routes, promising substantial improvements in both the economic and operational metrics of pharmaceutical intermediate manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of key intermediates such as (S)-2-amino-3-methylsulfonyl-benzyl phenylpropionate hydrochloride (Compound VIII) has relied heavily on the use of L-3-bromophenylalanine as the foundational chiral scaffold. As illustrated in the prior art reaction scheme, this traditional approach necessitates a cumbersome four-step sequence involving Boc protection, methyl sulfone functionalization, esterification, and final deprotection. The reliance on L-3-bromophenylalanine presents a significant bottleneck; this starting material is not only commercially expensive but also subjects the supply chain to volatility associated with natural amino acid sourcing. Furthermore, the multi-step nature of the conventional route inherently accumulates impurities and reduces overall yield, as each transformation requires isolation, purification, and quality control checks. The introduction of the sulfone moiety in the later stages of the traditional synthesis often requires harsh conditions or specialized reagents, further complicating the process safety profile and increasing the environmental footprint due to higher solvent and reagent consumption per kilogram of final product.
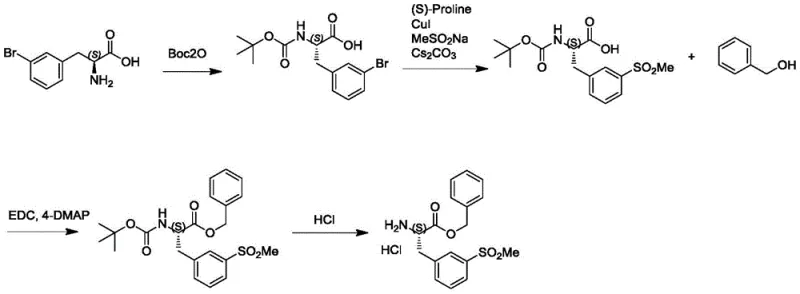
The Novel Approach
In stark contrast to the legacy methods, the invention disclosed in CN111471003B proposes a highly convergent and atom-economical strategy that drastically simplifies the synthetic landscape. The core innovation lies in the disconnection of the target molecule into two inexpensive fragments: a brominated sulfone derivative (Compound I or V) and a chiral iodo-alanine ester (Compound II or VI). By employing a Negishi cross-coupling reaction, these two fragments are joined in a single catalytic step to form the crucial carbon-carbon bond, effectively installing the aryl sulfone group onto the amino acid backbone with retention of configuration. This approach reduces the synthetic linear step count significantly, eliminating the need for late-stage functionalization of the aromatic ring. The use of cheap, commodity-grade chemicals like 3-bromophenyl methyl sulfone replaces the costly chiral amino acid starter, fundamentally altering the cost structure of the synthesis. Additionally, the final deprotection step is straightforward, yielding the target hydrochloride salt directly, which simplifies downstream processing and crystallization workflows.
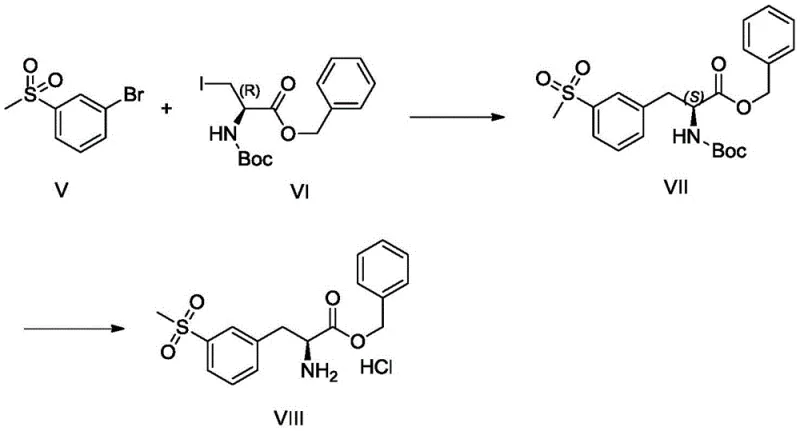
Mechanistic Insights into Pd-Catalyzed Negishi Coupling
The success of this novel route hinges on the efficient generation and utilization of an organozinc reagent under mild conditions. The mechanism initiates with the activation of metallic zinc powder, often facilitated by additives like methanesulfonic acid in polar aprotic solvents such as DMAC (N,N-dimethylacetamide). This activation step is critical for breaking the oxide layer on the zinc surface, allowing for the smooth insertion of zinc into the carbon-iodine bond of the N-Boc-3-iodo-L-alanine ester. The resulting organozinc species is highly nucleophilic yet sufficiently stable to be handled in situ. Upon introduction to the palladium catalytic cycle, the organozinc reagent undergoes transmetallation with the palladium(II) species formed from the oxidative addition of the aryl bromide. The choice of ligand, such as tri(o-methylphenyl)phosphine or tris(dibenzylideneacetone)dipalladium, plays a pivotal role in stabilizing the active catalytic species and preventing beta-hydride elimination, which could otherwise lead to racemization or side products. This careful tuning of the catalytic system ensures that the chiral center at the alpha-position of the amino acid remains intact throughout the coupling process, delivering the product with high enantiomeric excess.
Impurity control is another hallmark of this mechanistic design. By avoiding the harsh conditions required for direct sulfonation of the aromatic ring in the traditional route, the potential for regio-isomeric impurities is virtually eliminated. The coupling reaction is highly selective for the carbon-bromine bond, leaving other functional groups like the ester and the protected amine untouched. Post-reaction workup involves simple aqueous extraction and filtration, which effectively removes palladium residues and zinc salts. The final acid-mediated deprotection of the Boc group is clean and quantitative, precipitating the product as a hydrochloride salt which can be easily purified by recrystallization. Experimental data from the patent confirms that this mechanistic robustness translates to exceptional purity profiles, with multiple examples demonstrating assay values greater than 99.0% and often reaching 99.5% without the need for chromatographic purification, a feat that is rare for such complex chiral intermediates.
How to Synthesize (S)-2-Amino-3-Methylsulfonyl-Benzyl Phenylpropionate Efficiently
The practical implementation of this synthesis is designed for ease of operation in a standard GMP manufacturing facility. The process begins with the preparation of the organozinc reagent in a dedicated vessel, where temperature control is maintained between 10-20°C during the addition of the iodo-alanine derivative to ensure safety and reagent stability. This solution is then transferred to a second reactor containing the aryl bromide and the palladium catalyst system, where the coupling proceeds at elevated temperatures (70-80°C) to drive the reaction to completion within a short timeframe. Following the coupling, the reaction mixture is worked up by quenching with water and extracting with organic solvents like dichloromethane. The crude coupled product is then subjected to acidic conditions to remove the Boc protecting group, causing the final product to precipitate as a white solid. This streamlined workflow minimizes unit operations and solvent swaps, making it an ideal candidate for rapid technology transfer and scale-up.
- Activation of zinc powder with methanesulfonic acid in DMAC followed by the dropwise addition of N-Boc-3-iodo-L-alanine ester to generate the reactive organozinc reagent at controlled temperatures.
- Execution of the Negishi coupling reaction between the generated organozinc species and 3-bromophenyl methyl sulfone using a palladium catalyst and phosphorus ligand system.
- Final deprotection of the Boc group using acidic conditions (e.g., HCl in ethyl acetate) to isolate the target amino acid hydrochloride salt with high purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthesis route offers transformative benefits that extend far beyond simple chemistry. The shift from expensive, specialty chiral starting materials to commodity chemicals fundamentally de-risks the supply chain. By decoupling the production from the volatile market of protected amino acids, manufacturers can secure more stable pricing and ensure continuity of supply even during global shortages of biological feedstocks. The reduction in synthetic steps also correlates directly with a reduction in manufacturing lead time, allowing for faster response to market demand fluctuations. Furthermore, the high purity achieved directly from crystallization reduces the burden on quality control laboratories and minimizes the risk of batch failures due to out-of-specification impurities, thereby enhancing overall operational efficiency and reliability for the downstream drug product manufacturing.
- Cost Reduction in Manufacturing: The economic impact of replacing L-3-bromophenylalanine with 3-bromophenyl methyl sulfone cannot be overstated. The starting materials for the new route are significantly cheaper and more abundant, leading to a drastic reduction in the Bill of Materials (BOM) cost. Additionally, the缩短 of the synthetic route from four steps to essentially two major steps eliminates the costs associated with intermediate isolations, solvent recovery, and labor for each skipped unit operation. The high yield and purity observed in the examples suggest that waste generation is minimized, further lowering the cost of goods sold (COGS) through improved material efficiency and reduced waste disposal fees.
- Enhanced Supply Chain Reliability: Relying on commodity chemicals like zinc powder, aryl bromides, and simple esters creates a much more resilient supply chain compared to relying on complex, fermentation-derived amino acid derivatives. These raw materials are produced by a wide range of global chemical suppliers, reducing the risk of single-source dependency. The robustness of the Negishi coupling chemistry also means that the process is less sensitive to minor variations in raw material quality, providing a buffer against supply fluctuations. This reliability ensures that production schedules can be met consistently, preventing delays in the delivery of the final Active Pharmaceutical Ingredient (API) to patients.
- Scalability and Environmental Compliance: The patent explicitly demonstrates the scalability of this process with a successful run in a 50L reactor, producing over 700g of product with consistent quality. This proof of concept at pilot scale indicates that the chemistry translates well to larger industrial reactors without significant re-optimization. From an environmental perspective, the shorter route and higher atom economy result in a lower E-factor (mass of waste per mass of product). The avoidance of harsh sulfonation reagents and the use of recyclable solvents like DMAC and ethyl acetate align with modern green chemistry principles, facilitating easier regulatory approval and reducing the environmental compliance burden for the manufacturing site.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation, ensuring accuracy and relevance for process development teams evaluating this technology for adoption.
Q: What are the primary advantages of this Negishi coupling route over conventional methods?
A: The novel route utilizes significantly cheaper starting materials like 3-bromophenyl methyl sulfone instead of expensive L-3-bromophenylalanine, reducing the synthetic sequence from four steps to just two major transformations while maintaining high optical purity.
Q: What purity levels can be achieved with this manufacturing process?
A: Experimental data from the patent indicates that the final product, such as Compound VIII, can be consistently obtained with purity levels exceeding 99.0%, often reaching 99.5% after simple filtration and drying procedures.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the process has been successfully demonstrated in a 50L reactor setup, proving its scalability. The use of robust reagents and standard workup procedures facilitates easy adaptation to multi-kilogram or ton-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Sitagliptin Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust and scalable synthetic routes in the modern pharmaceutical landscape. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to full-scale manufacturing. Our state-of-the-art facilities are equipped to handle sensitive organometallic reactions like the Negishi coupling described in CN111471003B, with stringent purity specifications and rigorous QC labs dedicated to maintaining the highest standards of quality. We understand that the integrity of your supply chain depends on the reliability of your intermediate supplier, and we are committed to delivering consistent, high-quality batches that meet your exacting requirements.
We invite you to leverage our technical expertise to optimize your production costs and secure your supply of critical amino acid intermediates. Our team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume needs, demonstrating how the adoption of this novel route can impact your bottom line. Please contact our technical procurement team today to request specific COA data, route feasibility assessments, and a comprehensive proposal for your upcoming projects. Let us partner with you to bring life-saving medications to market faster and more efficiently.