Optimizing Elagolix Production: A Technical Analysis of Patent CN109810065B for Commercial Scale-Up
Optimizing Elagolix Production: A Technical Analysis of Patent CN109810065B for Commercial Scale-Up
The pharmaceutical landscape for treating endometriosis has been significantly shaped by the development of GnRH antagonists, with Elagolix standing out as a critical therapeutic agent. As demand for this active pharmaceutical ingredient (API) continues to rise, the efficiency of its synthetic route becomes a paramount concern for global supply chains. Patent CN109810065B, published in mid-2022, introduces a refined synthesis method that addresses long-standing challenges in yield and purity associated with earlier methodologies. This technical insight report analyzes the proprietary chemistry disclosed within this patent, focusing on its implications for R&D feasibility, procurement cost structures, and supply chain reliability. By shifting away from problematic polyalkylation pathways, this innovation offers a robust framework for the commercial scale-up of complex pharmaceutical intermediates. The core of this advancement lies in a strategic two-step transformation that converts a protected pyrimidine precursor directly into the target molecule with enhanced control over impurity profiles.
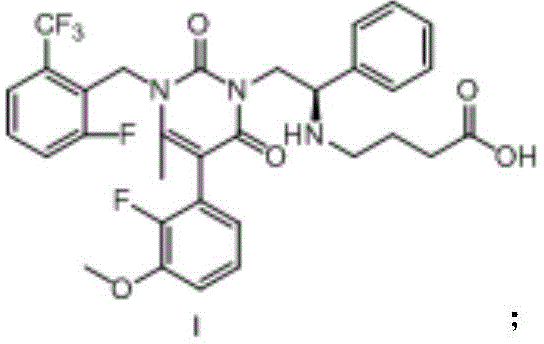
Understanding the molecular architecture of Elagolix is essential for appreciating the synthetic hurdles involved. The molecule features a complex uracil-derived core substituted with fluorinated aromatic rings and a specific amino acid side chain. Achieving the correct stereochemistry and functional group placement without generating structural analogs is the primary challenge for process chemists. The patent highlights that previous attempts often resulted in mixtures that were difficult to separate, thereby driving up the cost of goods sold (COGS) due to material loss during purification. The new approach detailed in CN109810065B provides a clear pathway to overcome these limitations, positioning it as a vital technology for any reliable API intermediate supplier aiming to capture market share in the reproductive health sector.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art, specifically referenced as US Patent WO2009062087, established the foundational chemistry for Elagolix but suffered from significant process inefficiencies that hindered industrial adoption. The traditional synthetic route was plagued by low overall yields, primarily due to unselective reaction pathways during the alkylation stages. A major technical bottleneck identified in the background of CN109810065B is the propensity for N-polyalkylation side reactions. When attempting to install the necessary side chains, the nucleophilic nitrogen atoms on the pyrimidine ring would often react multiple times with alkylating agents, creating a spectrum of over-alkylated byproducts. These impurities possess physicochemical properties very similar to the target molecule, making their removal via standard crystallization or chromatography extremely difficult and costly. Furthermore, the multi-step nature of the conventional route increased the cumulative material loss at each stage, exacerbating the economic burden on manufacturers.
From a supply chain perspective, the reliance on such inefficient chemistry translates to longer lead times and higher volatility in pricing. The need for extensive purification steps not only consumes valuable solvent and energy resources but also extends the batch cycle time, reducing the throughput of manufacturing facilities. For procurement managers, this means that sourcing Elagolix intermediates produced via legacy methods often comes with a premium price tag and less flexibility in delivery schedules. The environmental footprint is also larger due to the increased E-factor (mass of waste per mass of product), which is becoming an increasingly critical metric for compliance with green chemistry regulations in Europe and North America. Consequently, there is a pressing industry need to transition toward more atom-economical and selective synthetic strategies.
The Novel Approach
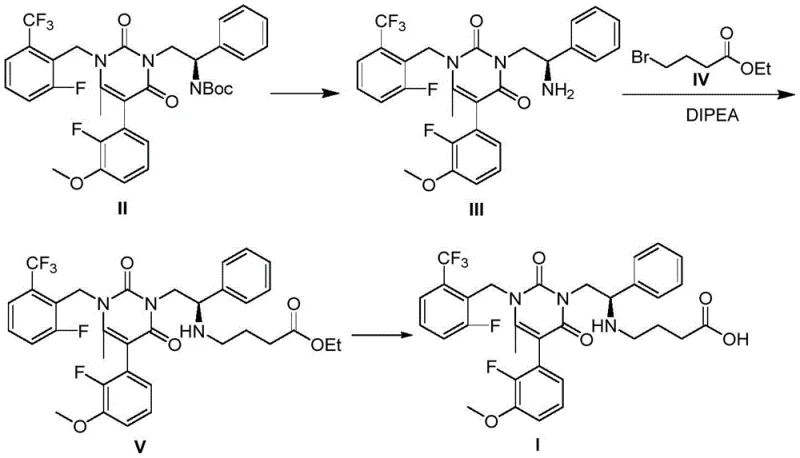
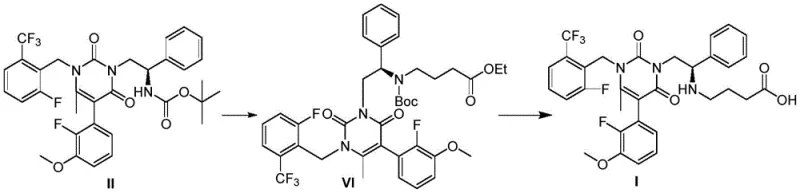
The invention disclosed in CN109810065B presents a paradigm shift by streamlining the synthesis into a highly efficient two-step sequence. The core innovation involves the N-alkylation of a specific tert-butyl carbamate precursor, designated as Formula II, with ethyl 4-bromobutyrate (Formula IV). This reaction is meticulously controlled to favor mono-alkylation, thereby drastically reducing the formation of polyalkylated impurities. The resulting intermediate, Formula VI, retains the necessary protecting groups to ensure stability during subsequent processing. The second step involves a concurrent deprotection strategy where both the N-Boc and O-Et groups are removed under basic conditions to yield the final carboxylic acid form of Elagolix (Formula I). This telescoped logic minimizes the number of isolation steps and maximizes the retention of chiral integrity throughout the process.
This novel approach offers substantial advantages in terms of process robustness and scalability. By utilizing commercially available starting materials like ethyl 4-bromobutyrate and standard phase transfer catalysts, the route eliminates the need for exotic or expensive reagents that often bottleneck supply chains. The reaction conditions are relatively mild, typically operating between 25°C and 90°C, which reduces the energy load on production reactors. Moreover, the workup procedures described involve standard aqueous washes and organic extractions, which are easily adaptable to existing infrastructure in multipurpose API plants. For a reliable agrochemical intermediate supplier or pharma partner, this translates to a lower barrier to entry for manufacturing and a more predictable production timeline. The ability to achieve high purity directly from the reaction mixture reduces the dependency on resource-intensive preparative HPLC, further driving down operational costs.
Mechanistic Insights into N-Alkylation and Deprotection
The success of this synthetic route hinges on the precise control of nucleophilic substitution dynamics during the first step. The reaction mechanism involves the displacement of the bromide ion from ethyl 4-bromobutyrate by the secondary amine nitrogen of the pyrimidine precursor. To facilitate this, the patent specifies the use of phase transfer catalysts such as tetrabutylammonium bromide (TBAB) or tetrabutylammonium iodide (TBAI). These catalysts play a crucial role in solubilizing the ionic species in the organic phase, thereby increasing the collision frequency between the nucleophile and the electrophile. The choice of base is equally critical; organic bases like DIPEA or pyridine are employed to scavenge the generated hydrogen bromide without promoting unwanted elimination reactions. The reaction is typically conducted in polar aprotic solvents such as DMF, DMAc, or 1,2-dichloroethane, which stabilize the transition state and enhance reaction kinetics. Maintaining the temperature at approximately 90°C ensures that the activation energy barrier is overcome efficiently, leading to conversion rates that support high isolated yields ranging from 59% to 78% across different examples.
Impurity control is inherently built into the mechanistic design of the second step. The deprotection of intermediate Formula VI is achieved using inorganic bases like potassium carbonate or cesium carbonate in a mixed solvent system of water and an organic co-solvent (e.g., THF or methanol). This hydrolytic environment selectively cleaves the ester and carbamate bonds while leaving the sensitive fluorinated aromatic rings and the chiral center intact. The use of mild heating (25-50°C) prevents racemization, which is a common risk in peptide-like syntheses. By avoiding harsh acidic conditions often used in Boc deprotection, the process minimizes the degradation of the urea linkage within the pyrimidine core. This selectivity ensures that the final product meets stringent purity specifications required for regulatory filing. The mechanistic elegance of this route lies in its orthogonality; the conditions chosen for alkylation do not interfere with the protecting groups, and the conditions for deprotection do not compromise the newly formed carbon-nitrogen bond.
How to Synthesize Elagolix Efficiently
Implementing this synthesis protocol requires careful attention to stoichiometry and reaction monitoring to replicate the high yields reported in the patent examples. The process begins with the dissolution of the Boc-protected precursor in a suitable organic solvent, followed by the sequential addition of the alkylating agent, base, and catalyst. It is imperative to maintain anhydrous conditions during the alkylation phase to prevent hydrolysis of the ethyl ester before the intended deprotection step. Once the intermediate is formed and isolated, the subsequent hydrolysis must be monitored via HPLC to ensure complete conversion without over-exposure to basic conditions which could lead to ring opening. The detailed standardized synthesis steps below outline the specific parameters for temperature, time, and reagent ratios that have been validated to produce high-quality Elagolix suitable for downstream formulation.
- Perform N-alkylation of the tert-butyl carbamate precursor (Formula II) with ethyl 4-bromobutyrate (Formula IV) using a phase transfer catalyst and base in an organic solvent at elevated temperatures.
- Isolate the intermediate ethyl ester (Formula VI) through aqueous workup and chromatographic purification.
- Execute simultaneous N-Boc and O-Et deprotection on the intermediate using a strong base in a mixed solvent system to yield the final Elagolix acid (Formula I).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of the synthesis method described in CN109810065B represents a strategic opportunity to optimize the cost structure of Elagolix manufacturing. The primary economic driver here is the significant reduction in processing complexity. By condensing the synthesis into fewer steps with higher individual yields, the overall material throughput is improved, meaning less raw material is required to produce the same amount of final API. This efficiency directly correlates to cost reduction in pharmaceutical intermediates manufacturing, as the consumption of expensive solvents and reagents is minimized. Furthermore, the avoidance of transition metal catalysts eliminates the need for costly and time-consuming metal scavenging processes, which are often required to meet strict residual metal limits in drug substances. This simplification of the downstream processing train allows for faster batch turnover and reduced utility consumption.
- Cost Reduction in Manufacturing: The elimination of complex purification steps and the use of commodity chemicals like ethyl 4-bromobutyrate and potassium carbonate significantly lowers the variable cost per kilogram. The high selectivity of the N-alkylation step reduces the generation of waste byproducts, thereby lowering waste disposal costs and improving the overall atom economy of the process. Additionally, the ability to use standard reactor materials without the need for specialized corrosion-resistant linings (often required for harsh acidic deprotections) reduces capital expenditure requirements for new production lines.
- Enhanced Supply Chain Reliability: The reliance on widely available, off-the-shelf reagents mitigates the risk of supply disruptions associated with custom-synthesized building blocks. Since the raw materials are commodity chemicals produced by multiple global vendors, procurement teams can leverage competitive bidding to secure favorable pricing and ensure continuity of supply. The robustness of the reaction conditions also means that the process is less sensitive to minor variations in raw material quality, providing a buffer against supply chain volatility and ensuring consistent production output even when sourcing from different suppliers.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing unit operations that are standard in the fine chemical industry. The reduced solvent usage and higher yields contribute to a lower environmental footprint, aligning with corporate sustainability goals and regulatory pressures to reduce chemical waste. The absence of heavy metals simplifies the environmental permitting process for new manufacturing sites and reduces the liability associated with hazardous waste management. This makes the technology particularly attractive for companies looking to expand capacity in regions with strict environmental regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the experimental data and claims presented in patent CN109810065B, providing a factual basis for decision-making. Understanding these nuances is critical for R&D teams evaluating technology transfer and for commercial teams negotiating supply agreements. The clarity provided here aims to de-risk the adoption of this methodology by addressing potential concerns about reproducibility, safety, and regulatory compliance upfront.
Q: How does this new synthesis route improve upon the prior art described in WO2009062087?
A: The conventional route suffers from low yields and significant N-polyalkylation side reactions which introduce difficult-to-remove impurities. The novel method described in CN109810065B utilizes a specific N-alkylation strategy followed by a streamlined deprotection step, effectively suppressing polyalkylation and significantly enhancing the purity profile of the final API intermediate.
Q: What are the critical reaction conditions for the N-alkylation step?
A: The process requires heating the reaction mixture to approximately 90°C or reflux conditions for 6 to 32 hours. Critical reagents include a phase transfer catalyst such as TBAB or TBAI (0.1-0.2 equiv) and a base like DIPEA or pyridine, utilizing solvents such as DMF, DMAc, or 1,2-dichloroethane to ensure optimal conversion rates.
Q: Is this synthesis method suitable for large-scale commercial production?
A: Yes, the method is highly scalable as it relies on commercially available raw materials and avoids complex transition metal catalysis. The two-step sequence simplifies the operational workflow, reducing the number of unit operations required and facilitating easier purification, which is essential for cost-effective manufacturing at the 100 MT scale.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Elagolix Supplier
As the global demand for endometriosis treatments continues to grow, securing a stable and high-quality supply of Elagolix intermediates is more critical than ever. NINGBO INNO PHARMCHEM stands at the forefront of this sector, leveraging advanced synthetic methodologies like the one described in CN109810065B to deliver superior value to our partners. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements without compromising on quality. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch meets the highest industry standards. Our commitment to technical excellence ensures that we can navigate the complexities of fluorinated chemistry and chiral synthesis with precision.
We invite you to collaborate with us to optimize your supply chain for Elagolix and related pharmaceutical intermediates. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific production needs. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us demonstrate how our innovative approach to synthesis can drive efficiency and reliability in your manufacturing operations, ensuring a steady flow of high-purity materials for your critical drug development programs.