Scalable Manufacturing of High-Purity 3'-O-Methoxyethyl Nucleoside Intermediates
The rapid advancement of genomic medicine has placed antisense oligonucleotide (ASO) drugs at the forefront of therapeutic innovation, driven by their superior specificity and reduced side effect profiles compared to traditional small molecules. Central to the efficacy of second-generation ASOs is the incorporation of chemically modified nucleosides, specifically 3'-O-methoxyethyl nucleosides, which enhance binding affinity and nuclease resistance. However, the industrial realization of these critical building blocks has historically been hindered by significant synthetic challenges, particularly the difficulty in controlling regioselectivity during the modification of the ribose ring. Patent CN108424432B discloses a groundbreaking preparation method that overcomes these barriers by utilizing a modified allose starting material to achieve high-purity 3'-O-methoxyethyl nucleosides without the generation of difficult-to-separate 2'-isomers. This technical breakthrough represents a paradigm shift for reliable pharmaceutical intermediate supplier networks seeking to secure the supply chain for next-generation genetic medicines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for 3'-O-modified nucleosides often rely on the direct alkylation of unprotected or partially protected ribose derivatives, a process fraught with thermodynamic pitfalls. In these conventional scenarios, the hydroxyl groups at the 2' and 3' positions possess similar reactivities, leading to the inevitable formation of a complex mixture of 2'-O-methoxyethyl and 3'-O-methoxyethyl isomers. Separating these structural analogs is notoriously difficult due to their nearly identical physical and chemical properties, typically necessitating the use of preparative high-performance liquid chromatography (prep-HPLC) or repeated, yield-depleting crystallization cycles. From a manufacturing perspective, reliance on prep-HPLC is economically unsustainable for large-scale production, as it severely limits throughput, increases solvent consumption, and drastically reduces overall process yield. Furthermore, the inability to completely remove these isomers poses a significant regulatory risk, as impurities in oligonucleotide drugs can lead to unpredictable off-target effects or immunogenic responses, making the conventional approach unsuitable for cost reduction in pharmaceutical intermediate manufacturing.
The Novel Approach
The innovative methodology described in the patent circumvents these regioselectivity issues entirely by employing a chiral pool strategy starting from 1,2:5,6-diacetone-D-allofuranose. Instead of attempting to differentiate between similar hydroxyls on a ribose ring, the synthesis builds the correct stereochemistry from the outset through a highly selective alkylation at the 3-position of the allofuranose scaffold. This is followed by a strategic oxidative cleavage of the 5,6-diol and subsequent reduction, which effectively shortens the carbon chain to generate the desired ribose configuration with the methoxyethyl group locked exclusively at the 3'-position. By utilizing benzoyl groups for robust protection of the remaining hydroxyls, the process ensures that the subsequent glycosylation reaction proceeds with high stereoselectivity to form the beta-anomer. This route eliminates the formation of 2'-isomers at the source, thereby removing the need for complex chromatographic purification and enabling the production of high-purity intermediates through simple crystallization, significantly enhancing the commercial scale-up of complex nucleoside analogs.

The mechanistic elegance of this synthesis lies in its precise control over stereochemistry and functional group manipulation, which is critical for R&D directors focused on impurity profiles. The initial alkylation step utilizes potassium hydroxide in dimethyl sulfoxide (DMSO) to selectively deprotonate the 3-hydroxyl group of the diacetone allose, facilitated by the steric environment of the acetonide protecting groups. This ensures that the methoxyethyl moiety is introduced with absolute regioselectivity. Following this, the selective removal of the 5,6-acetonide group exposes the vicinal diol, which is then subjected to periodate oxidation to cleave the carbon-carbon bond, generating an aldehyde intermediate. Subsequent reduction with sodium borohydride converts this aldehyde into the primary alcohol, completing the transformation from the allose skeleton to the ribose framework. The final glycosylation step employs trimethylsilyl trifluoromethanesulfonate (TMSOTf) as a potent Lewis acid catalyst to activate the anomeric center of the fully benzoylated sugar intermediate. This activation promotes the nucleophilic attack by the silylated nitrogenous base (such as thymine or cytosine derivatives), driving the formation of the N-glycosidic bond with high beta-selectivity. The use of silylated bases, prepared in situ using N,O-bis(trimethylsilyl)acetamide (BSA), enhances the nucleophilicity of the base while maintaining anhydrous conditions essential for preventing hydrolysis of the activated sugar.
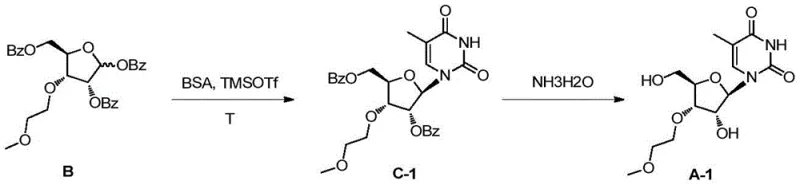
Impurity control is inherently built into this pathway through the stability of the benzoyl protecting groups and the crystallinity of the intermediates. Unlike acetyl groups which can migrate under acidic or basic conditions leading to isomerization, benzoyl groups provide robust protection that withstands the harsh conditions of glycosylation and subsequent deprotection. The final deprotection step utilizes aqueous ammonia, which cleanly removes the benzoyl esters without affecting the ether linkage of the methoxyethyl group or the integrity of the nucleobase. This orthogonal stability ensures that the final product stream contains minimal degradation by-products. Furthermore, the ability to crystallize the final nucleoside from ethanol indicates a high degree of lattice energy and purity, allowing for the rejection of trace organic impurities and residual solvents. For quality assurance teams, this means the process consistently delivers material meeting stringent purity specifications (>99%) required for clinical-grade oligonucleotide synthesis, minimizing the burden on downstream analytical testing and batch release protocols.
How to Synthesize 3'-O-Methoxyethyl Nucleosides Efficiently
The synthesis of these high-value intermediates follows a logical progression from cheap commodity chemicals to complex chiral building blocks, designed for operational simplicity and safety. The process begins with the alkylation of the sugar scaffold, followed by oxidative restructuring and finally, coupling with the nucleobase. Each step utilizes standard reactor equipment and commonly available reagents, avoiding the need for specialized high-pressure or cryogenic setups. The detailed standardized synthesis steps see the guide below, which outlines the specific reaction conditions, stoichiometry, and workup procedures optimized for maximum yield and purity.
- Alkylation of 1,2: 5,6-diacetone-D-allofuranose with 2-chloroethyl methyl ether using KOH/DMSO to form the 3-O-methoxyethyl intermediate.
- Selective deprotection and oxidative cleavage of the 5,6-diol followed by reduction to generate the ribose configuration.
- Benzoyl protection of hydroxyl groups followed by condensation with silylated bases using TMSOTf catalyst to yield the final nucleoside.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition from chromatography-dependent methods to this crystallization-based route offers profound economic and logistical benefits. The elimination of preparative HPLC not only reduces capital expenditure on specialized equipment but also drastically lowers operating costs associated with solvent consumption, column replacement, and extended processing times. By relying on crystallization for purification, the process becomes inherently more scalable, allowing for batch sizes to be increased linearly without the bottlenecks typical of chromatographic separations. This scalability ensures a stable and continuous supply of critical raw materials, reducing the risk of production delays for downstream oligonucleotide manufacturers. Additionally, the use of inexpensive starting materials like diacetone allose and standard reagents such as benzoyl chloride and potassium hydroxide insulates the supply chain from the volatility associated with exotic catalysts or proprietary reagents.
- Cost Reduction in Manufacturing: The most significant financial advantage stems from the complete avoidance of preparative chromatography, which is traditionally the most expensive unit operation in nucleoside synthesis. By achieving high purity through crystallization, the process eliminates the massive solvent volumes and low throughput associated with HPLC, leading to substantial cost savings in both raw materials and waste disposal. Furthermore, the high overall yield of the route, driven by the high selectivity of each step, maximizes the output per kilogram of starting material, effectively lowering the cost of goods sold (COGS) for the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route contributes directly to supply chain resilience. Because the chemistry relies on well-understood transformations and stable intermediates, the risk of batch failure due to sensitive reaction conditions is minimized. The ability to produce multiple nucleoside variants (thymine, cytosine, adenine, guanine derivatives) from a common advanced intermediate (Compound B) allows for flexible manufacturing scheduling. This modularity enables suppliers to respond quickly to fluctuating demands for specific ASO sequences without needing to maintain separate, dedicated production lines for each nucleoside type, thereby optimizing inventory management and reducing lead times for high-purity nucleoside intermediates.
- Scalability and Environmental Compliance: From an environmental and safety perspective, the process is designed for green manufacturing principles. The reagents used are relatively benign compared to heavy metal catalysts often found in cross-coupling reactions, simplifying wastewater treatment and regulatory compliance. The concentration of waste streams is lower due to the higher efficiency of crystallization compared to chromatography, reducing the environmental footprint of the manufacturing site. Moreover, the absence of special equipment requirements means that the technology can be transferred to diverse manufacturing sites globally, facilitating regional production hubs that reduce shipping distances and enhance the security of supply for global pharmaceutical partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and process descriptions within the patent, providing clarity on the feasibility and performance of the method for potential licensees or contract manufacturing partners.
Q: How does this method avoid the formation of 2'-O-isomers?
A: The process utilizes a specific starting material (modified allose) and a strategic protection/deprotection sequence that inherently directs substitution to the 3'-position, eliminating the thermodynamic mixture of 2'/3' isomers common in direct alkylation methods.
Q: Is preparative HPLC required for purification?
A: No. A key advantage of this patented route is that the final products can be purified to >99% purity via simple crystallization from ethanol, removing the need for expensive and low-yield preparative chromatography.
Q: What bases are compatible with this synthesis route?
A: The method is versatile and has been successfully demonstrated with thymine, 5-methylcytosine, adenine, and 2,6-diaminopurine, covering the essential bases required for antisense oligonucleotide drug development.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3'-O-Methoxyethyl Nucleoside Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of antisense therapies depends on the availability of ultra-high purity nucleoside building blocks produced via robust, scalable routes. Our technical team has thoroughly analyzed the patented methodology for 3'-O-methoxyethyl nucleosides and confirmed its viability for industrial application. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to pilot plant and finally to full-scale manufacturing is seamless. Our facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, ensuring that every batch of intermediate meets the exacting standards required for GMP oligonucleotide synthesis.
We invite pharmaceutical developers and procurement leaders to collaborate with us to leverage this advanced synthesis technology for your pipeline projects. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us today to obtain specific COA data for our reference standards and to discuss route feasibility assessments for your custom nucleoside needs, ensuring your supply chain is optimized for both cost and quality.